鈮酸鉀鈉基無鉛壓電陶瓷的高壓電活性研究進展*
2020-07-04 07:35:08邢潔譚智鄭婷吳家剛肖定全朱建國
物理學報 2020年12期
邢潔 譚智 鄭婷 吳家剛 肖定全 朱建國
(四川大學材料科學與工程學院, 成都 610064)
(2020 年2 月25日收到; 2020 年3 月20日收到修改稿)
以Pb(Zr1—xTix)O3 (PZT)為代表的鉛基壓電陶瓷因為具有良好的壓電性能和機電耦合性能已被廣泛應用于科技、工業、軍事以及日常生活中. 但是, PZT基陶瓷中Pb的含量超過了60% (質量比), 在生產、使用及廢棄處理過程中都會給人類生態環境造成嚴重損害. 因此, 發展無鉛壓電陶瓷已成為世界壓電陶瓷研究的熱點之一. 鈮酸鉀鈉 (K0.5Na0.5)NbO3 (KNN)無鉛壓電陶瓷因為具有較為優異的壓電性能以及較高的居里溫度, 被認為是最可能取代鉛基壓電陶瓷的材料體系之一. 經過研究者們的努力工作, 改性后的KNN基無鉛壓電陶瓷壓電性能已經接近或超過了某些鉛基壓電陶瓷的性能. 本文綜合介紹了具有高壓電活性的KNN基無鉛壓電陶瓷國內外的研究進展, 重點闡述了高性能鈮酸鉀鈉基無鉛壓電陶瓷制備工藝及相關理論基礎的研究進展, 并就今后鈮酸鉀鈉基無鉛壓電陶瓷研究發展的方向及前景提出建議.
1 引 言
壓電材料能夠將機械能和電能互相轉換, 已經在各種電子器件中得到應用[1,2]. 常見的壓電材料有壓電單晶、壓電陶瓷、壓電高分子材料以及壓電復合材料等. 其中, 壓電陶瓷因為具有制備工藝簡單、成本低廉、性能優異、性能可控性強等特點, 已經被廣泛應用于科技、工業、軍事以及日常生活中,對整個國民經濟的發展有著深刻的影響.
隨著現代科學技術的進步和發展, 壓電陶瓷從最初的BaTiO3(BT)體系發展到以Pb(Zr1—xTix)O3(PZT)為代表的鉛基壓電陶瓷體系. 由于在準同型相界(MPB)附近的PZT基陶瓷具有非常優異的綜合電學性能, 因此目前應用最為廣泛. 但是,PZT基陶瓷中Pb的含量約為原料總量的60%,而Pb的毒性以及易揮發性使鉛基壓電陶瓷在生產、使用及廢棄后處理過程中都會給生態環境和人類自身健康造成嚴重損害[3,4]. 近年來, 世界各國都立法禁止使用含鉛的電子材料, 如歐盟頒布執行的《關于限制在電子電器設備中使用某些有害成分的指令》(Restriction on the Use of Hazardous Substancces), 日本通過的《家用電子產品回收法案》, 我國信息產業部出臺的《電子信息產品污染防治管理辦理》等. 因此, 發展環境友好的無鉛壓電陶瓷成為當前國際壓電材料研究的熱點之一[3,5-8].
目前, 主要的無鉛壓電陶瓷可分為以下幾種結構: 鈣鈦礦結構、鎢青銅結構、鉍層狀結構. 因為鈣鈦礦結構壓電陶瓷的壓電性能優異, 制備工藝與傳統鉛基陶瓷工藝兼容, 是目前研究最廣泛的一大類無鉛壓電陶瓷. 常見的鈣鈦礦結構無鉛壓電陶瓷主要包括BT基、鈦酸鉍鈉(Bi0.5Na0.5TiO3, BNT)基和鈮酸鉀鈉(K0.5Na0.5NbO3, KNN)基等.
作為最早發現的BT壓電陶瓷, 具有介電常數高、機電耦合系數大、介電損耗較小等特點, 但是因為該類陶瓷的居里溫度較低(TC≈ 120 ℃), 其物理性能的溫度穩定性差, 并且燒結溫度高(大于1350 ℃), 因此, 需要大幅度提升其壓電性能來滿足實際應用的需求. 目前, 更多地是利用BT陶瓷具有高介電性能的特點而作為介質材料進行應用.
BNT陶瓷具有較高居里溫度(TC≈ 320 ℃)、高剩余極化強度(Pr≈ 38 μC/cm2)、較好的壓電性能以及較低的燒結溫度[9], 但BNT陶瓷室溫下矯頑場很大(Ec≈ 73 kV/cm)、去極化溫度較低(約為100 ℃)等, 從而限制了該類陶瓷的進一步應用.
KNN基陶瓷具有高居里溫度(TC≈ 420 ℃)和較好的壓電性能, 但是純的KNN陶瓷存在高溫下K, Na等元素易揮發、工藝敏感性強、難以通過傳統陶瓷工藝實現致密化等問題, 長期以來研究進展緩慢. 2004年, Saito等[10]通過化學摻雜改性,利用反應模板晶粒生長(RTGG)法制備的KNN基織構陶瓷性能達到d33≈ 416 pC/N, 機電耦合系數kp≈ 0.61,TC≈ 253 ℃. 2018 年, Li等[11]采用織構化工藝, 加入3 mol%的NaNbO3作為模板制備的0.96(K0.5Na0.5)(Nb0.965Sb0.035)O3-0.01Ca ZrO3-0.03(Bi0.5K0.5)HfO3織構陶瓷體系, 其d33高達 700 pC/N,kp≈ 0.76,TC≈ 242 ℃. 2019 年, Tao等[12]采用固相合成法, 制備的(0.96—x)K0.48Na0.52Nb0.95Sb0.05O3-0.04Bi0.5(Na0.82K0.18)0.5HfO3-0.4%Fe2O3-xAgSbO3非織構陶瓷, 其d33可達到 (650 ±20) pC/N,TC≈ 180 ℃. 這些陶瓷的性能接近甚至超過了PZT4的性能, 表明KNN基陶瓷是最有可能取代鉛基壓電陶瓷的無鉛壓電材料體系之一.
本文基于國際最新研究結果和研究工作, 綜合分析并簡要介紹了KNN基無鉛壓電陶瓷高壓電活性的國內外的研究, 特別是高性能KNN基無鉛壓電陶瓷、制備工藝優化與相關理論基礎的研究進展, 并對今后的研究方向進行了展望.
2 高性能KNN基無鉛壓電陶瓷
采用固相燒結法制備的KNN陶瓷的d33大約是80 pC/N[13], 與實際應用的鉛基陶瓷壓電性能差距較大. 為了進一步提升KNN基無鉛壓電陶瓷的壓電性能, 研究者們對KNN基陶瓷進行了詳細的研究, 研究的重點主要集中在相界調控、提升陶瓷體系壓電性能以及綜合性能的優化與溫度穩定性等方面.
2.1 高壓電性能
2.1.1 相界構建
與PZT基壓電陶瓷類似, KxNa1—xNbO3陶瓷是由鐵電體KNbO3和反鐵電體NaNbO3形成的二元固溶體. 隨著溫度的變化, KNN陶瓷會在—123 ℃ 時經歷三方相 (R)-正交相 (O)轉變, 在210 ℃ 時發生正交相-四方相 (T)轉變, 在 410 ℃下發生四方相-立方相(C)轉變[8]. 利用離子摻雜或取代、添加第二組元或多組元化合物等方法, 可將KNN基無鉛壓電陶瓷的相轉變溫度移動到室溫附近, 構建室溫下的新型相界. 在新型相界區域,因為多相共存, 有更多可能的極化狀態, 不同相之間的各向異性減小, 電疇更易偏轉, 從而使KNN基無鉛壓電陶瓷的性能得到提高[8,14].
在KNN基中添加一定的離子(Li+, Ag+, Sb5+,Ta5+)或 Bi0.5(K/Na/Li)0.5TiO3, (Ba/Ca/Sr)TiO3等鈣鈦礦結構化合物, 可以提升KNN基陶瓷中四方相的比例, 把TO-T由200 ℃左右調節至室溫附近, 使KNN基陶瓷在室溫下從單一的正交相結構轉變為O-T相共存, 構建出室溫下的O-T相界,提升KNN基陶瓷的壓電性能. 表1 歸納了一些構建室溫下O-T相界的KNN基陶瓷體系的研究進展[15-23].
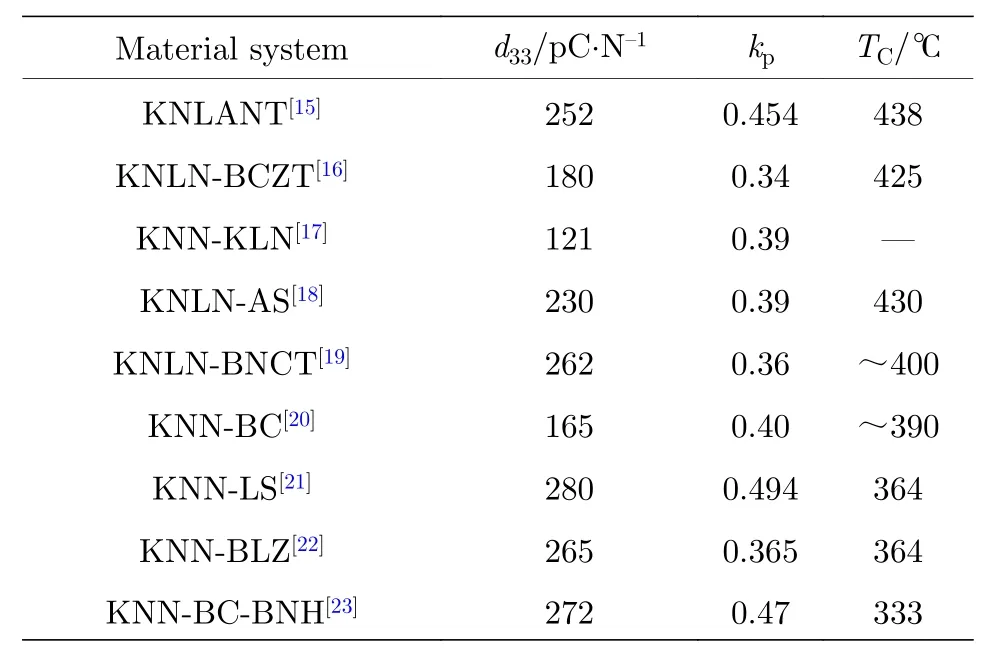
表 1 室溫下具有O-T相界的KNN壓電陶瓷性能Table 1. Properties of KNN ceramics with O-T phase boundary at room temperature.
從表1可以看出, 可以通過一定的離子摻雜或者多組元復合, 提升KNN基陶瓷的壓電性能, 尤其是添加了Li+的陶瓷體系, 能夠在獲得較高壓電性能的同時保持較高的居里溫度.
通過構建室溫下的O-T相界, 可以提升KNN基陶瓷的壓電活性, 但是相對于鉛基陶瓷仍有一定的差距. 在KNN基陶瓷中降低TO-T的同時, 添加一定的離子(Sb5+, Ta5+)或Bi0.5(K/Na/Li/Ag)0.5(Zr/Hf)O3, (Ba/Ca/Mg)ZrO3, Bi(Sc/Co/Fe)O3等化合物, 可以提升KNN基陶瓷中三方相的比例,把TR-O由—123 ℃左右調節至室溫附近, 在室溫下構建出三方-正交-四方(R-O-T)或三方-四方(RT)新型相界. 其中R-O-T新型相界是同時調控TO-T和TR-O至室溫附近, 使其室溫下同時存在R,O, T三相. R-T新型相界是進一步壓縮陶瓷體系的TO-T和TR-O之間的溫區, 使其在室溫附近重疊, 形成新的相界, 室溫下存在R相和T相. 多相共存時, 相與相之間存在較低的能量勢壘, 促進極化翻轉, 優化KNN基陶瓷的電學性能. 表2和表3分別列出了室溫下構建R-O-T[12,24-34]和R-T[35-47]相界的部分研究成果.

表 2 室溫下具有R-O-T相界的KNN壓電陶瓷的性能Table 2. Properties of KNN ceramics with R-O-T phase boundary at room temperature.
從表1—3可以看出, 相較于O-T相界, 具有R-O-T/R-T相界的KNN基無鉛壓電陶瓷的電學性能提升程度更大, 表明相界類型對其壓電活性影響很大. 相較于只降低TO-T構建室溫下O-T相界, 在KNN陶瓷中構建R-O-T/R-T相界則更加復雜, 需要引入多種添加離子或者化合物才能實現. 值得注意的是, 由于KNN陶瓷對組分和工藝的敏感性以及摻雜離子的相互影響, 在KNN基陶瓷中構建新型相界需要進行組分和工藝的精細調控.
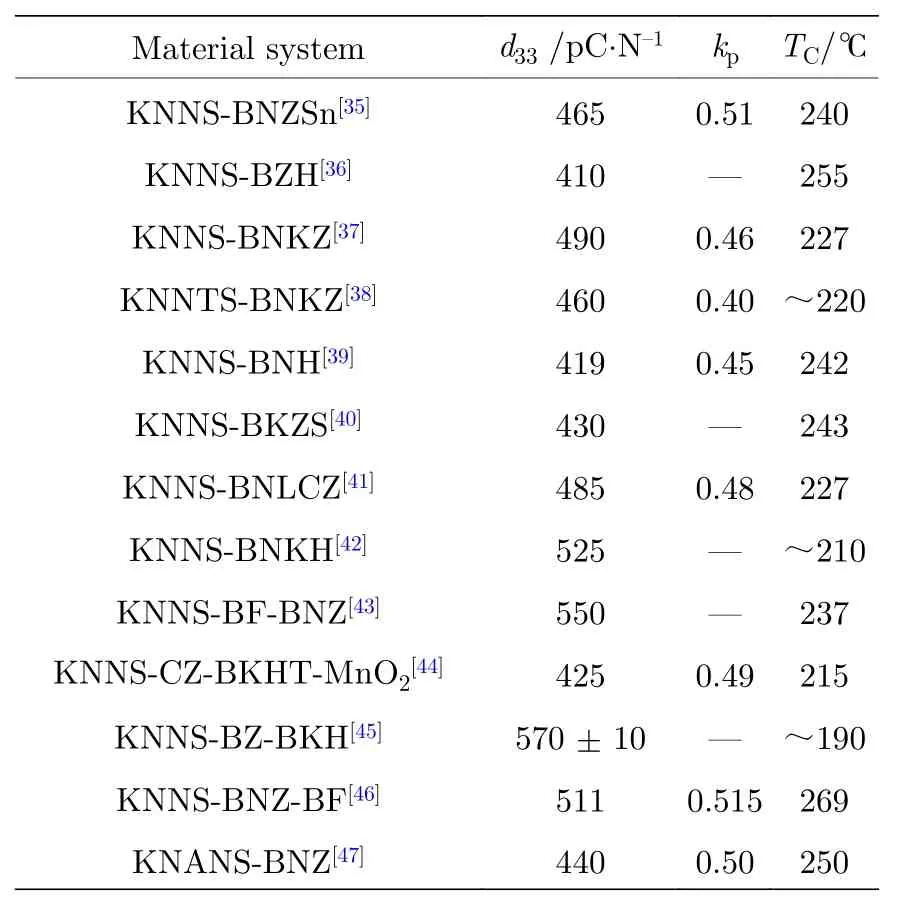
表 3 室溫下具有R-T相界的KNN壓電陶瓷的性能Table 3. Properties of KNN ceramics with R-T phase boundary at room temperature.
圖1(a)給出了KNN基無鉛壓電陶瓷的壓電常數d33的歷史演變圖. 2014年Wang等[37]利用固相燒結法合成的 (1—x)(K1—yNay)(Nb1—zSbz)O3-xBi0.5(Na1—wKw)0.5ZrO3非織構陶瓷, 室溫下具有R-T 相界, 陶瓷的d33≈ 490 pC/N,TC≈ 190 ℃.隨后該團隊合成的KNNS-BZ-BKH非織構陶瓷,其d33高達(570 ± 10) pC/N,TC≈ 190 ℃; KNNSBF-BNZ 非織構陶瓷[43], 其d33≈550 pC/N,TC≈237 ℃. 2018年, Li等[11]采用織構化工藝制備的KNNS-CZ-BKH織構陶瓷, 其d33≈ 700 pC/N,kp≈0.76. Liu等[48]報道了具有高壓電性能兼具優異的溫度穩定性的KNN-BZ-BNH-MnO2非織構陶瓷, 其> 600 pm/V. 2019 年 Tao 等[12]報道了KNNS-BNKH-0.4%Fe2O3-xAgSbO3非織構陶瓷, 具有 R-O-T 相界, 其d33≈(650 ± 20) pC/N,TC≈ 180 ℃. Zhou等[34]通過兩步燒結法制備了(K,Na)(Nb,Sb)O3-(Bi,Na)ZrO3-BaZrO3非織構陶瓷, 并獲得了d33≈ 610 pC/N,kp≈ 0.58,k33≈0.69的高壓電性能. 經過改性后的KNN基無鉛壓電陶瓷壓電性能與鉛基壓電陶瓷的性能差距逐漸縮小, 如圖1(b)所示. 總的來說, 通過新型相界的構建, KNN基陶瓷體系的壓電性能已經可以接近甚至超過部分鉛基陶瓷的水平, 在部分壓電器件和應用中具有取代PZT基陶瓷體系的潛力.
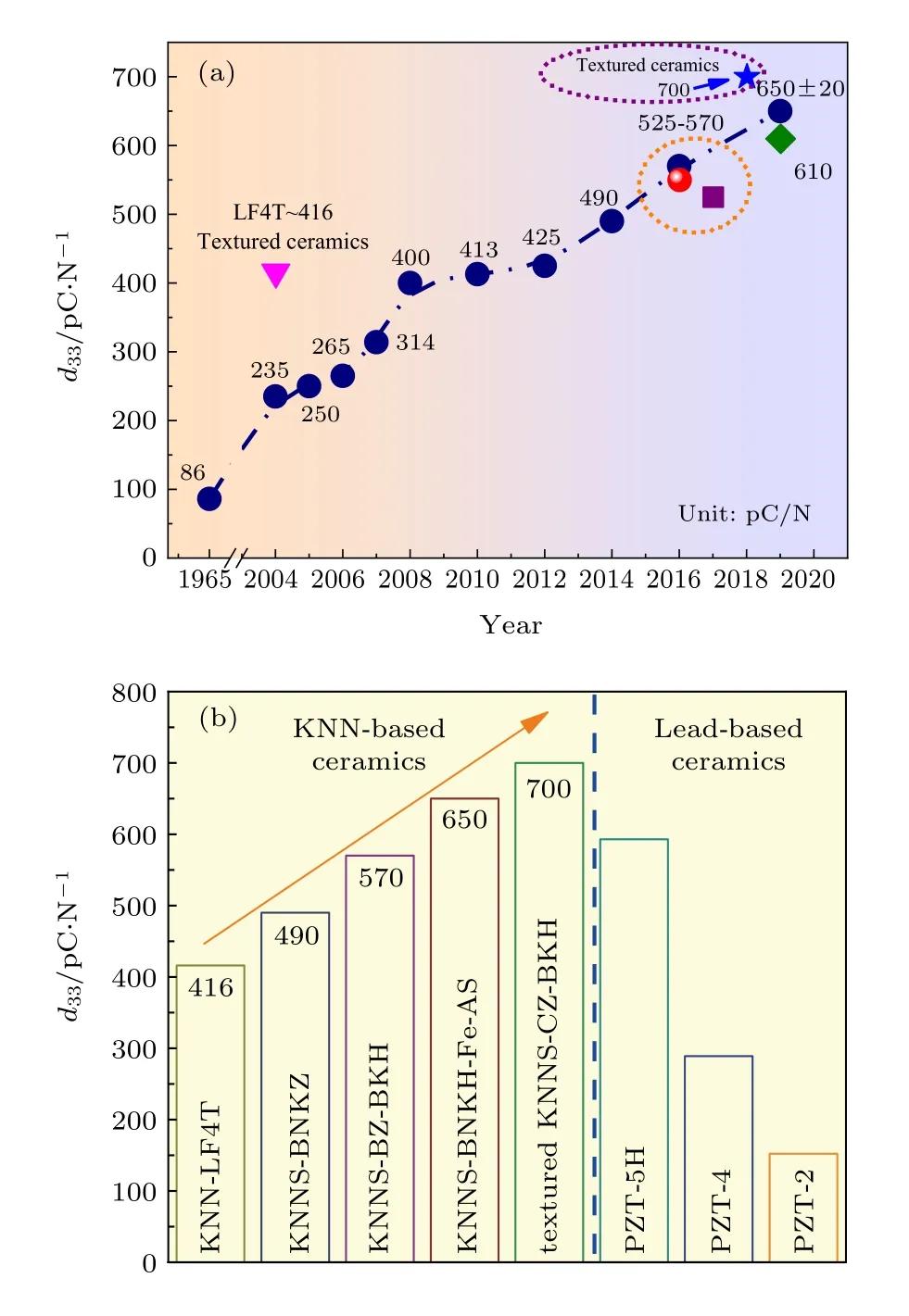
圖 1 (a) KNN基無鉛壓電陶瓷d33的歷史演變圖; (b) KNN基無鉛壓電陶瓷與鉛基陶瓷 d33 對比圖 [11,12,34,37,43,45]Fig. 1. (a) Historical evolution in d33 values of KNN-based ceramics as a function of time; (b) comparison of d33 values among KNN-based ceramics and PZT materials[11,12,34,37,43,45].
2.1.2 疇結構調控
鐵電材料在降溫過程中, 在低于居里溫度時,鐵電體從順電相轉變為鐵電相, 其晶體結構從高對稱性轉變為低對稱性. 晶體的正負中心不再重合而形成自發極化, 并且在同一個鐵電相中, 自發極化存在多個等效的自發極化方向. 對于鐵電晶體來說, 由于自發極化使其正負兩端產生束縛電荷, 束縛電荷產生的電場與極化方向相反, 會在鐵電晶體中產生退極化場. 為了降低退極化場形成的靜電能以及晶格畸變產生的應變能, 在鐵電單晶或者陶瓷中會出現許多小的區域, 每個區域內的晶胞自發極化方向相同, 而相鄰區域內自發極化方向取向不同. 自發極化方向一致的小區域稱為電疇. T相中的自發極化方向為方向, 存在6個等效的自發極化方向, O相的自發極化方向沿方向,有12個自發極化方向, R相的自發極化方向沿方向, 有8個等效自發極化方向. 未加外場時,各電疇的自發極化方向不同, 相互抵消, 使其宏觀極化強度為零. 在外場作用下, 電疇的自發極化矢量發生翻轉. 電疇和電疇之間的邊界稱為疇壁. 根據疇壁兩側的極化方向夾角不同, KNN基壓電陶瓷 T相含有 90°疇和 180°疇, O相晶體為 90°疇、180°疇、60°疇和 120°疇, R 相則具有 71°疇、109°疇以及180°疇.
壓電陶瓷的壓電響應分為本征與非本征貢獻兩部分, 其中本征貢獻主要取決于晶格畸變, 而非本征貢獻與電疇運動有關, 包括疇壁振動、電疇翻轉、疇壁運動. 因此, 除了構建相界外, 疇結構調控對于提升KNN基無鉛壓電陶瓷的壓電性能至關重要.
電子顯微技術是常用的電疇觀測方法, 包括透射掃描電子顯微鏡(TEM)、壓電力顯微鏡(PFM)以及掃描電子顯微鏡(SEM)等. 將合成后的KNN基無鉛壓電陶瓷樣品進行減薄處理后可以通過TEM進行電疇結構的觀測[49-51], 在測量疇結構尺寸的同時還可以利用選區電子衍射進行特定區域的結構分析[49]. 使用原位透射電鏡觀測, 則能夠觀察外電場作用下電疇結構的變化[52]. 因為不同極性電疇被酸腐蝕的程度不同, 通過該特點可以采用SEM直接觀察腐蝕后的KNN基陶瓷電疇結構. 文獻[53-55]利用混合酸溶液(濃度為37%的鹽酸與40%氫氟酸溶液體系比1∶1)對KNN基陶瓷進行腐蝕, 并通過SEM觀測了其相應的疇結構.López-Juárez等[56]使用 48 vol.% 的氫氟酸腐蝕(K0.5Na0.5)NbO3陶瓷體系后, 觀測到正交相的陶瓷中90°疇和180°疇結構. 壓電力顯微鏡(PFM)因為分辨率高、對樣品要求低, 是一種研究疇結構的重要檢測方法, 被廣泛應用在KNN基無鉛壓電陶瓷中[57,58]. 其中, 利用原位PFM中電場、溫度場的變化可以判斷KNN基無鉛壓電陶瓷中疇的翻轉難易程度以及溫度穩定性[59,60].
通過電子顯微技術可以在高壓電性能、多相共存的KNN基陶瓷中同時觀察到條紋疇以及亞微米級不規則的疇結構. 圖2(a)—(c)給出了在室溫下不含相界, O-T, R-O-T, R-T相界的KNN基陶瓷的疇結構. 未摻雜的K0.5Na0.5NbO3陶瓷, 其疇結構多為魚骨狀、水痕狀, 其中魚骨狀疇結構對應90°疇, 水痕狀疇結構對應 180°疇[61]. K0.5Na0.5NbO3陶瓷在室溫下為正交相, 其90°疇結構尺寸為 0.5—1 μm 寬以及 3—7 μm 長, 180°疇的尺寸為 3—10 μm 寬, 20—40 μm 長[56]. 通過構建室溫下的新型相界, KNN陶瓷體系中的疇結構形狀或者尺寸發生改變. 如圖2(b)和圖2(c)所示, 對于KNN基陶瓷中構建了室溫下的O-T/R-O-T/RT 相界時[34,43,62,63], 可以利用 TEM 或者 SEM 觀察到條紋疇或者不規則形狀的復雜疇結構, 并且疇結構的尺寸下降到亞微米級或者納米級. 構建室溫下新型相界可以使KNN陶瓷具有更多極化方向. 相對于室溫下為O相的KNN陶瓷, 位于多相共存區域的KNN陶瓷含有更多的極化狀態和晶體取向度, 不同相之間的各向異性減小, 因此疇的翻轉和移動比單相陶瓷更容易[8,14]. 有研究表明, 新型相界施加一定的外加電場后, 除了發生電疇翻轉外還會產生電誘導相變, 誘導相為單斜相(M), 極化矢量通過中間誘導相更易翻轉, 從而增強陶瓷的壓電性能[49,64].

圖 2 (a)正交相(K0.5Na0.5NbO3陶瓷[56,61]); (b)室溫下O-T相界(KNNL-BZ-BNT陶瓷體系[62], KNNSL-BNZ-BZ-MnO2陶瓷體系[63]);(c)室溫下R-T/R-O-T相界KNN基陶瓷的疇結構(KNNS-BF-BNZ陶瓷體系[43], KNNS-BNZ-BZ陶瓷體系[34])Fig. 2. Domain structures of KNN-based ceramcis with different phase boundaries at room temperature: (a) Orthorhombic(K0.5Na0.5NbO3 ceramics[56,61]); (b) O-T phase boundaries (KNNL-BZ-BNT ceramics[62], KNNSL-BNZ-BZ-MnO2 ceramics[63]); (c) R-T/R-O-T phase boundaries (KNNS-BF-BNZ ceramics[43], KNNS-BNZ-BZ ceramics[34]).
在KNN基無鉛壓電陶瓷中進行多組元摻雜, 可以顯著地降低各元素的擴散速率, 破壞晶體的長程有序, 從而容易形成納米級尺寸的電疇. 例如, Sun 等[52]發現 (K0.48Na0.52)(Nb0.955Sb0.045)O3-0.01SrZrO3陶瓷體系未摻入(Bi0.5Ag0.5)ZrO3前的疇結構尺寸為0.2—1 μm, 在摻入(Bi0.5Ag0.5)ZrO3后的陶瓷體系中同時觀察到了30—65 nm, 65—160 nm的條狀疇以及30—45 nm尺寸的納米疇. 該陶瓷體系摻雜后的d33≈ 487 pC/N. Liu等[48]構建的KNNS-BZ-BNH-MnO2體系具有10—100 nm尺寸的疇結構, 其壓電系數為600 pm/V. Zheng等[42]通過TEM研究發現在KNN陶瓷中摻入Sb和Bi(Na, K)HfO3可以使陶瓷具有寬度為10—30 nm, 長度為 100—300 nm尺寸的納米疇, 從而提高該陶瓷的壓電性能(d33≈ 525 pC/N). Tao等[12]制成的高壓電性能(d33≈ (650 ± 20) pC/N)的KNN基陶瓷體系的疇結構約為2 nm的納米疇. Zhou等[34]獲得的KNN基陶瓷壓電性能d33≈610 pC/N, 疇結構尺寸為50—70 nm. 表4列舉了一些高性能KNN基無鉛壓電陶瓷壓電常數與疇結構尺寸[12,34,42,46,48,51,52,54,57,65-70]. 從表 4 可以看出,疇結構尺寸與陶瓷壓電性能的優異密切相關, 尤其是納米尺寸的疇結構對陶瓷的高壓電性能起著重要的作用. 這是因為疇壁能與疇尺寸的平方根成正比[56], 納米尺度的疇結構在外電場作用下更容易翻轉, 從而對KNN陶瓷的電學性能起到增強作用.因此, 對疇結構進行調控可以提升KNN基體系的壓電性能.

表 4 KNN基無鉛壓電陶瓷壓電常數與疇結構尺寸Table 4. Piezoelectric constant of KNN ceramics with domain size.
2.2 溫度穩定性
在KNN基無鉛壓電陶瓷中添加三方或四方誘導物構建室溫下新型相界, 其本質是將KNN基陶瓷的相變點移動至室溫附近, 獲得高的壓電性能. 目前, KNN基無鉛壓電陶瓷的壓電性能已經可以滿足一部分壓電器件及應用的性能要求. 但是, 與PZT鉛基陶瓷材料中的準同型相界不同,KNN基陶瓷中構建的新型相界為多晶型相界(PPB), 不僅有組分依賴性, 還有溫度依賴性. 當溫度上升或者下降時, KNN基無鉛壓電陶瓷會發生相變恢復至單相結構, 從而導致該陶瓷體系壓電性能下降, 因此, 在小于TC的溫度范圍內存在另一個多晶型相變是KNN壓電陶瓷溫度穩定性差的本質原因. 因此, 為了能夠解決無鉛壓電陶瓷的應用問題, 還需要考慮KNN基無鉛壓電陶瓷的高居里溫度以及良好的溫度穩定性.
2.2.1 居里溫度
壓電陶瓷中壓電性能的提升往往伴隨著該體系居里溫度的下降, 如圖3所示[12,15-47]. 壓電材料可以在大約1/2TC—2/3TC的溫度范圍內安全使用. 因此, 在構建室溫下新型相界的同時, 應該考慮其實際應用的溫度范圍, 調控壓電性能與居里溫度之間的關系, 發展同時具有高壓電性能以及高居里溫度的KNN基無鉛壓電陶瓷體系, 這將對無鉛壓電陶瓷的發展具有極大的推動作用.
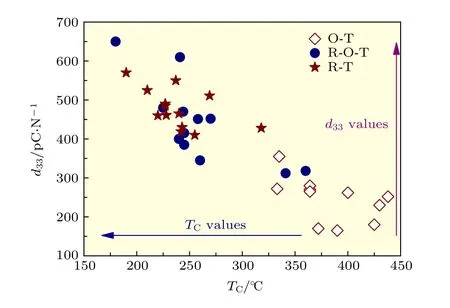
圖 3 KNN基無鉛壓電陶瓷d33與TC對比圖[12,15-47]Fig. 3. Comparison of d33 and TC values of KNN-based ceramics[12,15-47].
表5列舉了部分兼具高壓電性能高居里溫度的 KNN 陶瓷體系[24,57,71-81]. 其中 Jiang 等[74]通過在KNN基陶瓷中添加BiAlO3組元, 可以有效提升陶瓷的電學性能, 保持高居里溫度, 該陶瓷體系d33≈ 355 pC/N,TC≈ 335 ℃. 另外, 在 KNN 基陶瓷中添加BiGaO3[24], BiInO3[57]等組元也可以使陶瓷同時具有高d33和高TC.
在KNN中如果設計陶瓷體系的A位堿金屬為非化學計量比, 可以通過彌補高溫下的堿金屬揮發提高陶瓷體系的致密度, 從而提升KNN基壓電陶瓷的壓電性能[82-84], 而非化學計量比的Nb5+對KNN基壓電陶瓷的影響研究報道較少. 本課題組通過調節B位Nb5+非化學計量比的量來對KNNSBNZ陶瓷相界進行調控[70]. 通過該陶瓷體系的微觀形貌表征以及致密度的測試發現, 適量的過量Nb5+能夠形成A位空位, 加速原子的擴散, 從而使陶瓷致密度增加, 最終提高陶瓷的TC. 并且在不降低TC的前提下, 構建室溫下的R-T相界提升KNN陶瓷的壓電性能, 從而獲得既具有壓電性能又具有高TC的KNN基壓電陶瓷, 該陶瓷體系獲得的綜合性能如下:d33= 421 pC/N,kp= 0.48,TC= 256 ℃.
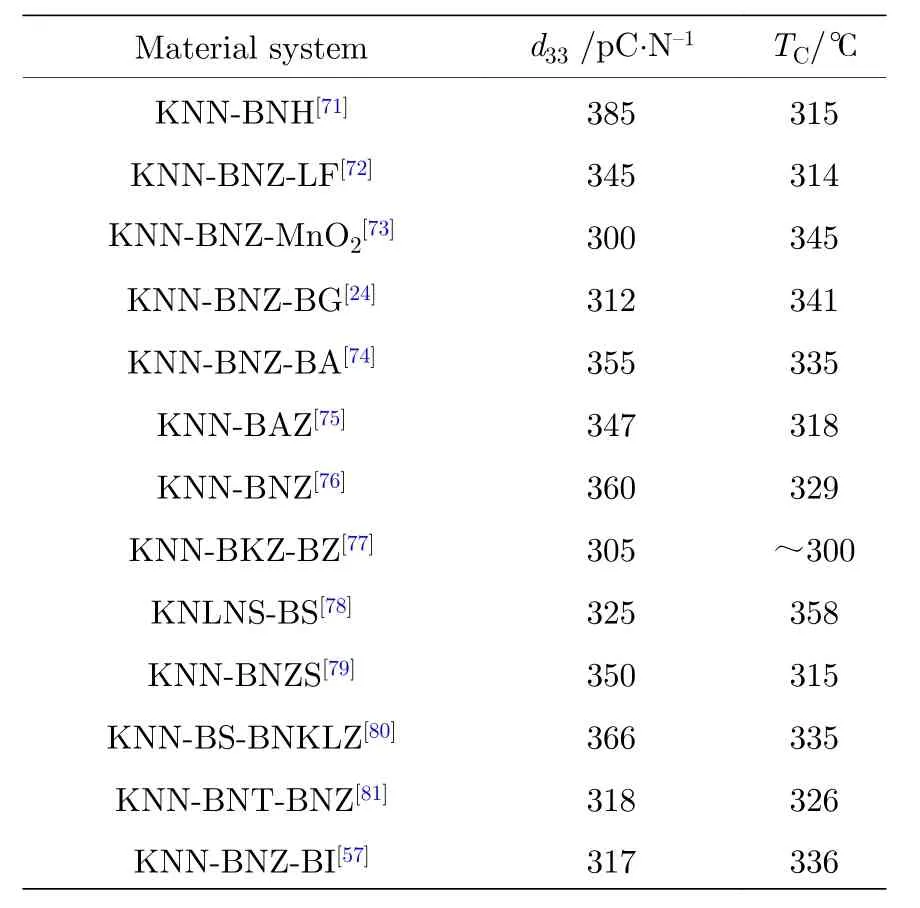
表 5 同時具有高壓電性能和高居里溫度的KNN陶瓷體系Table 5. The KNN-based ceramics with high piezoelectric constant and high Curie temperature.
2.2.2 溫度穩定性
陶瓷的溫度穩定性對KNN基陶瓷能否規模化應用至關重要. 研究者們通過多種方法增加KNN基無鉛壓電陶瓷的溫度穩定性. 將相變溫度移動至室溫以下能夠有效地改善KNN基陶瓷的溫度穩定性. Zhang等[85]在KNN-LS陶瓷體系中添加 CaTiO3, 將TO-T移動到室溫以下, 使其溫度穩定性增強, 但壓電性能大幅度下降.
構建彌散的新型相界, 在室溫附近形成一個溫度變化范圍比較大的新型相界, 能夠在增加陶瓷體系壓電性能的同時增加陶瓷體系的溫度穩定性.Yao等[86]通過在KNN基陶瓷中添加CaZrO3組元, 使KNN基陶瓷的O-T相變變得彌散, 延長相變的溫度范圍, 從而提升陶瓷的溫度穩定性, 使得該陶瓷體系的從室溫到140 ℃的溫度范圍內基本保持不變. 在KNN-BNZ陶瓷中添加LaFeO3移動TO-T, 使其O-T相界變得彌散, 在一定摻雜范圍內, 該陶瓷體系在室溫下為O-T兩相共存, 隨著LaFeO3摻雜量的增加, T相含量逐漸增加, 該陶瓷體系的在20—100 ℃溫度范圍內的變化量小于8%[72]. Zhou等[65]通過在KNN中添加可以同時提升TR-O和TO-T的 BaZrO3, (Bi, Na)ZrO3組元, 該陶瓷具有d33≈ 300 pC/N,kp≈ 0.56,k33≈ 0.69的高壓電性能, 該陶瓷體系因為彌散的R-O相變而在—30—100 ℃溫度范圍內具有優異的溫度穩定性. Yao等[86]通過原位變溫變電場同步輻射XRD, 變化外加電場強度, 發現了CaZrO3摻雜的KNN基無鉛壓電陶瓷體系中存在高電場誘導彌散型相變, 該陶瓷體系在高電場下獲得了更為優異的溫度穩定性.
與非織構化的KNN基陶瓷相比, KNN基陶瓷的織構化也可以有效地改善陶瓷的溫度穩定性. 相對于非織構化的LF4陶瓷在160 ℃的應變變化量為30%, 織構化的 LF4T陶瓷在160 ℃時的應變變化量為6.5%[10]. Li等[66]制備的織構化KNNS-CZ-BKH陶瓷體系, 通過加入NaNbO3模板, 獲得了納米尺寸的電疇結構, 因此在溫度為200 ℃ 時,d33值仍有室溫d33的 70%, 同時,x=0.03和0.04組分在室溫到150 ℃溫度范圍內應變的變化值小于20%.
KNN基無鉛壓電陶瓷中構建的新型相界類型是與陶瓷的溫度穩定性有關. Zhao等[60]通過對KNN基陶瓷構建不同相界, 并對比各種相界的溫度穩定性. 對比 R-O, O-T, R-O-T 相界, 發現具有R-T相界的KNN陶瓷體系具有優異的電學性能以及最好的溫度穩定性.
研究者們還發現調控晶粒尺寸或者疇結構也可以增加KNN基陶瓷體系的溫度穩定性. Cen等[51]認為KNN陶瓷體系的晶粒尺寸可以影響陶瓷的壓電性能以及溫度穩定性, 通過探討不同燒結溫度下制備的KNN基陶瓷體系的相結構以及晶粒尺寸, 發現在1180 ℃燒結溫度下的陶瓷具有適宜的晶粒尺寸. 晶粒尺寸的適當增長不僅能夠改變該陶瓷體系的相結構, 并增加了疇結構的尺寸, 從而降低了鐵電相到順電相相轉變溫度的弛豫性, 以此增加了該陶瓷體系的溫度穩定性. 1180 ℃燒結溫度下制備的陶瓷具有優異的壓電性能, 其=482 pm/V, 在120 ℃時仍具有良好的溫度穩定性.Feng等[49]通過在KNN基陶瓷引入SrZrO3設計并制備了KNNT-BNZ-SZ體系. 該陶瓷體系通過在A位中摻入 Sr2+,B位摻入 Ta5+和 Zr4+, 破壞了鐵電相的長程有序, 從而得到小尺寸的疇結構,增強溫度穩定性. 該陶瓷體系在x= 1.0%時獲得了= 400 pm/V,TC= 335 ℃的優異綜合性能, 該組分陶瓷在180 ℃以及190 ℃時壓電性能仍可以保持室溫下壓電性能的90%和85%. 表6中列舉了一些具有良好溫度穩定性的KNN基陶瓷[36,42,49,51,65,69,72,86-92].
雖然通過摻雜改性或者結構優化可以在一定程度上提升KNN基無鉛壓電陶瓷的溫度穩定性,然而與PZT基鉛基壓電陶瓷相比還有一定的差距, 并且如何從結構上解決KNN基無鉛壓電陶瓷的溫度敏感性還需要進一步的研究.
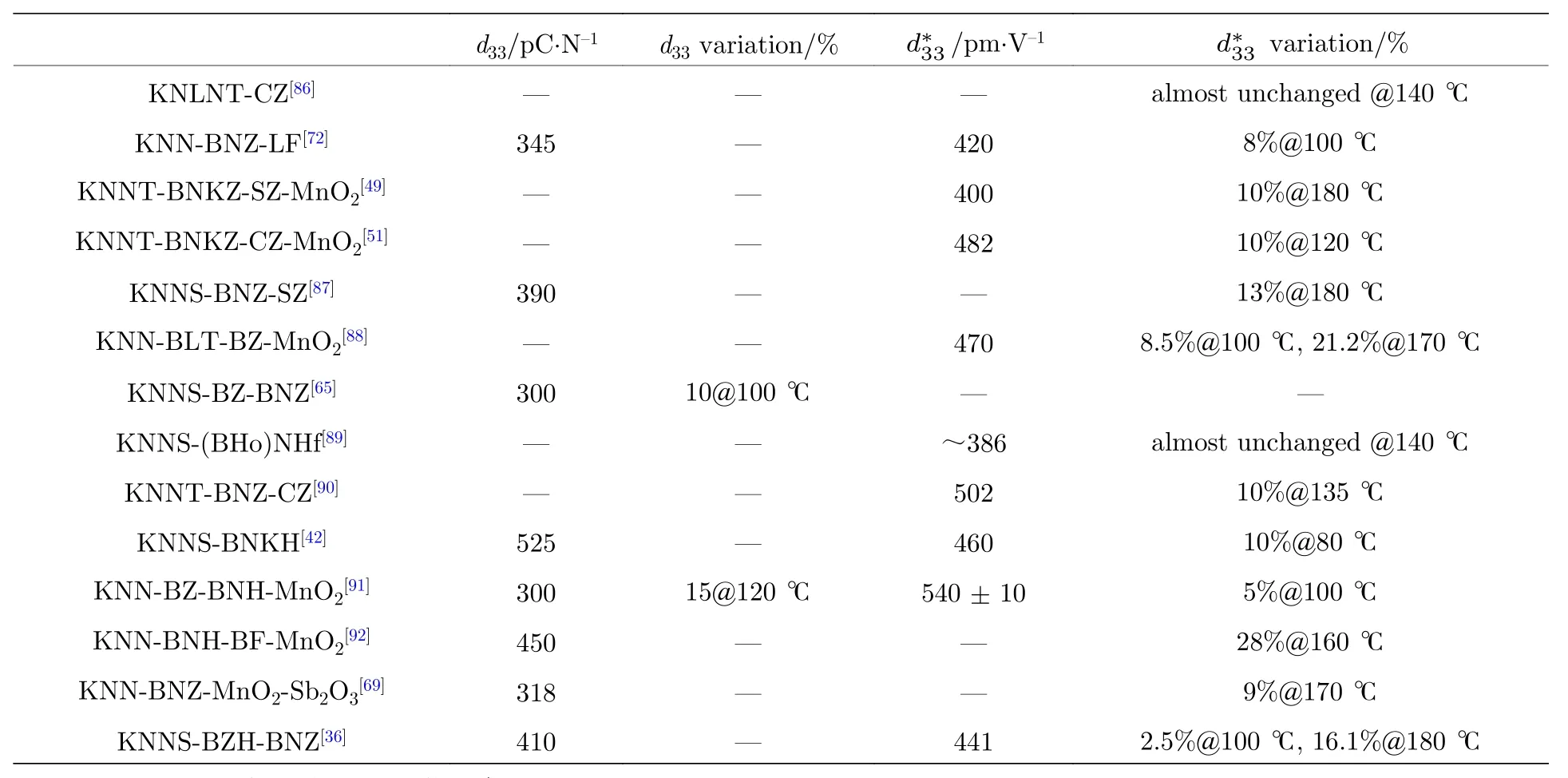
表 6 溫度穩定性高的KNN陶瓷體系的壓電常數以及變化量Table 6. Comparison of piezoelectric constant and variation among KNN-based ceramics.
3 制備方法
考慮到陶瓷的制備成本, 在陶瓷實際工程應用過程中常采取固相燒結法. 因為KNN基無鉛壓電陶瓷的燒結溫區較為狹窄, 高溫燒結過程中堿金屬元素易揮發等問題, 制備的KNN基陶瓷致密性較差, 從而影響陶瓷體系的電學性能. 改善粉體制備、燒結工藝、發展新的KNN基無鉛壓電陶瓷制備方法獲得了廣泛的關注.
3.1 粉體制備
盡管傳統固相法合成陶瓷粉體具有成本低、制備工藝簡單的優點, 但是該方法制備的粉體容易出現成分不均勻, 顆粒尺寸大, 不利于后續燒結工藝.除了固相法之外, 合成KNN粉體的主要方法有水熱合成法[93-96]、熔鹽法[97,98]、共沉淀法[99]、噴霧熱解法[100]等.
水熱合成法可以在處于低溫及高壓的水熱釜中一次性合成相應的超細粉體, 該方法對環境友好, 能夠精確控制粉體形貌及化學組成, 因此可以通過優化粉體微觀結構而改善KNN基陶瓷的性能[101]. Takeshi等[102]通過水熱法合成了尺寸小于1 μm 的 KNbO3, NaNbO3粉體, 制 備 了相應的KNN陶瓷體系. Liu等[94]通過水熱法分別合成KNbO3, NaNbO3以及 LiSbO3粉體, 通過優化粉體配比, 制備了 (1—x)K0.5Na0.5NbO3-xLiSbO3陶瓷體系, 獲得了d33= 183 pC/N, 機械品質因數Qm=99.83以及kp= 0.33的陶瓷體系.
熔鹽法制備工藝簡單, 能夠通過調控原料與鹽的比例, 精確控制合成納米粉體的形貌, 改善陶瓷的性能[97]. Li等[98]采用熔鹽法制備了KxNa1—xNbO3粉體, 通過調控原料與鹽的比例為1∶3以及合成溫度, 獲得了平均尺寸為1.5 μm的立方晶粒KNN粉體, 并在獲得的粉體中添加1 mol% ZnO為燒結助劑, 制備了性能為d33= 120 pC/N,TC= 406 ℃,Qm= 126以及kp= 0.302的KNN陶瓷體系.
共沉淀法可以直接通過化學反應獲得所需粉體, 并且制備的粉體顆粒尺寸均勻. Kumar等[99]采用共沉淀方法制備了K0.5Na0.5NbO3陶瓷, 該陶瓷具有高致密度以及增強的電學性能.
噴霧熱解法制備的粉體純度高, 分布均勻, 同時工藝簡單, 能夠連續化生產, 并且該方法所使用的水溶液前驅體成本低, 對環境友好, 有廣闊的工業應用前景[100]. Haugen等[100]通過噴霧熱解法制備了平均粒徑為130 nm的KNN粉體, 將該粉體進行常規燒結后制備出95%密度的KNN陶瓷,該陶瓷體系具有= 180 pm/V的性能.
3.2 燒結工藝
燒結是陶瓷坯體致密化的重要工藝, 傳統固相燒結具有工藝簡單、成本低等優點, 但是因為高溫燒結時K, Na元素易揮發, 該燒結工藝制備的陶瓷樣品難以致密. 優化燒結工藝對獲得高性能KNN陶瓷體系是十分重要的. 目前, KNN陶瓷的燒結工藝除了傳統固相燒結外, 還有二步燒結法[34,67,103]、熱壓燒結[104]、放電等離子體燒結[105]、微波輔助燒結[106]、冷燒結輔助燒結法[107,108]等.
兩步燒結法是先升溫至一個較高的溫度, 保溫一定時間后, 降低到一個較低的燒結溫度長時間進行保溫后隨爐冷卻, 這種方法能夠充分有效地提升KNN陶瓷的致密度, 從而增強陶瓷的電學性能[8].Liu等[67]通過兩步燒結法(快速升溫至1180 ℃,保溫 1 min后, 快速降溫至 1075 ℃, 在該溫度下保溫 7 h, 并隨爐冷卻), 制備的 0.963(K0.48Na0.52)(Nb0.955Sb0.045)O3-0.037(Bi0.50Na0.50)HfO3陶瓷的d33≈ 512 pC/N,kp≈ 0.54. 該課題組還通過兩步燒結法制備了(0.96—x)(K0.48Na0.52)(Nb0.96Sb0.04)O3-0.04(Bi0.50Na0.50)ZrO3-xBaZrO3陶 瓷 體 系 , 獲得了d33≈ 610 pC/N,kp≈ 0.58,k33≈ 0.69 的高壓電性能[34].
熱壓燒結法是在高溫燒結的同時對樣品進行加壓, 從而提高陶瓷致密度. Yu等[109]通過熱壓法制備的K0.5Na0.5NbO3-0.5 mol%Al2O3陶瓷體系,其d33為 137 pC/N,為 212 pm/V. 盡管該方法能夠提升陶瓷電學性能, 但該方法設備昂貴、成本較高, 不利于大型規模生產.
放電等離子體燒結法是一種低溫快速燒結方法. 該方法因為燒結溫度低, 能夠快速完成燒結過程, 抑制堿金屬元素的揮發, 從而提高陶瓷致密度,提高陶瓷壓電性能. Li等[110]采用該方法制備了K0.5Na0.5NbO3陶瓷, 該陶瓷的密度為 4.47 g/cm3,為理論密度的99%, 壓電性能為148 pC/N. 但是該方法同樣存在燒結設備昂貴、不利于工業化生產的缺點.
微波燒結法是利用微波加熱進行陶瓷燒結的方法. 該方法具有加熱時間短、能量利用率高(80%—90%)、安全清潔不污染等優點[106]. Feizpour等[106]利用微波燒結在短時間內制備的KNN陶瓷與傳統燒結法制備的陶瓷性能相匹配. 但是該方法存在升溫速率難以精確控制的弱點.
冷燒結輔助燒結法是一種將陶瓷粉體與去離子水、酒精或者溶液充分混合, 將混合后粉體在一定壓力下進行熱處理, 之后再進行燒結或者熱處理的一種方法. 該方法有助于提高陶瓷致密度,降低陶瓷燒結溫度. Ma等[107]采用該方法制備了K0.5Na0.5NbO3陶瓷, 將 K0.5Na0.5NbO3陶瓷粉體與不同質量百分比的去離子水進行混合, 在350 MPa,120 ℃下保溫30 min壓制成型, 在120 ℃下繼續保溫6 h去除殘余水后在1070—1145 ℃燒結4 h,最終獲得d33≈ 131 pC/N的KNN陶瓷. Chi等[108]將K0.5Na0.5NbO3陶瓷粉體與NaCl水溶液進行混合, 在 450 MPa下壓制成型, 在 120 ℃下烘干后8 h后, 900—1100 ℃下燒結3 h. 該陶瓷的壓電常數為d33≈ 115 pC/N,kp≈ 0.32,TC≈ 448 ℃.但是該方法在提升壓電性能方面還需要進一步研究.
燒結氣氛對陶瓷性能的影響也十分重要.Cen 等[111]在還原氣氛下 (PO2= 1 × 10—11MPa)燒結制備的CaZrO3摻雜KNN基無鉛壓電陶瓷,具有d33≈ 270 pC/N,≈ 490 pm/V, 同時該陶瓷體系在室溫至125 ℃溫度變化范圍內, 其變化量< 10%.
另外, 在KNN無鉛壓電陶瓷中進行不同金屬氧化物的摻雜在一定程度上都能夠有效地改善陶瓷的燒結特性, 提高陶瓷的致密度, 改善KNN基壓電陶瓷的相關性能, 而金屬氟化物摻雜的研究報道甚少. 2012年, Li等[112]通過在K0.5Na0.5Nb0.95Ta0.05O3陶瓷中添加1.0 wt% NaF作為燒結助劑,降低燒結溫度, 促進晶粒生長, 提高陶瓷致密度,相對于未添加NaF時陶瓷體系的d33≈ 110 pC/N,添加NaF后陶瓷體系的d33提升了40%左右, 達到153 pC/N. CuF2也是一種能夠有效降低KNN陶瓷燒結溫度, 提升陶瓷壓電性能的燒結助劑[113].Wu等[114]通過在KNN基陶瓷中摻雜ZnF2, 探討ZnF2對陶瓷微觀結構、電學性能以及溫度穩定性的影響. Liao等[115]通過合成K0.5Na0.5Nb0.996Cu0.01O3-xFx陶瓷體系, 探討了F-O取代對氧空位以及缺陷復合體的抑制作用, 通過進行F取代O, 陶瓷的d33≈103 pC/N,kp≈0.42、機械品質因子Qm≈1031.
3.3 制備工藝
陶瓷的織構化可以使陶瓷內部晶粒沿一定方向排列取向生長, 陶瓷在該方向上具有類似單晶的性能. 模板晶粒生長(TGG)法是在KNN粉料中按照一定比例加入片狀模板晶粒、黏結劑、增塑劑,配制成流延漿料后, 通過流延成型制備織構化的KNN基陶瓷. Li等[11]采用該方法制備了一系列KNN基陶瓷, 利用傳統固相法制備出KNN基陶瓷粉體, 并采用熔鹽法制備了NaNbO3模板, 坯體在600 ℃下預熱處理5 h后, 再采用兩步燒結法1190 ℃以及1090 ℃下燒結10 h制備, 制備出d33≈700 pC/N,kp≈ 0.76,TC≈ 242 ℃ 的 KNN 基織構陶瓷[11]. 研究發現, KNN基織構化壓電陶瓷的優異壓電性主要來源于三方面: 1)對于取向的織構陶瓷, 在極化電場下具有最優的工程疇結構; 2)原位同步輻射X射線衍射分析結果表明, 織構化陶瓷在外加電場下具有較大的晶格畸變且出現中間相, 這是壓電性能提高的本征貢獻;3)通過TEM和PFM的分析發現, 織構化陶瓷相比于同組分的隨機取向的陶瓷具有尺寸更小的電疇結構, 有利于電疇在外電場下的翻轉和疇壁的移動. 因此, 與普通陶瓷相比, 織構陶瓷的性能更為優異.
相較于傳統固相燒結法, 其他燒結方法或者工藝制備的KNN陶瓷能夠有效降低相應的粉體燒結溫度, 并且晶粒生長更為均勻, 從而較好地提高陶瓷的致密度. 但是這些方法工藝過程復雜, 生產成本較高, 難以實現大規模生產. 因此, 通過離子摻雜或組元取代方法提升KNN基陶瓷壓電性能仍然是目前研究的重點和主要方向.
4 理論基礎
通過研究相變的本質、相界構建機制、高壓電活性起源等, 可以為KNN基陶瓷的實驗制備以及器件應用提供理論依據.
4.1 鐵電相變
在相界處壓電陶瓷的介電和壓電系數等都會出現異常的增大, 目前絕大多數的高壓電材料都是在材料的相界處獲得的, 比如目前最常見的PZT陶瓷, 其性能最優的配方就處于三方-四方的準同型相界(MPB).
根據鈣鈦礦鐵電體的位移型相變理論, 鈣鈦礦鐵電體中的電極化形成主要來自于離子位移, 當溫度高于TC時, 材料處于順電相, 此時ABO3結構中A位離子位于晶胞頂角,B位離子位于體心, 而O離子在面心, 正負離子中心相互重合, 由此宏觀上不存在電極化. 而當溫度低于TC時, 離子相對于中心位置發生偏移, 正負電荷中心分離, 在晶胞內形成電偶極子, 由此造成了晶格結構的改變. 電極化的形成引起晶格結構發生畸變, 并改變晶格的對稱性, 晶格會沿極化方向明顯拉伸, 其余方向則相對壓縮, 鐵電相變因而形成. 位移型相變顯示出鐵電相中, 正負離子總是沿極化方向進行拉伸, 整體的晶體結構顯示出長程有序性.
鐵電相變的成因主要歸因于B位離子與O離子之間的強共價作用, 例如BaTiO3中, 按照位移型相變理論, 立方相的BaTiO3結構中Ti原子應該位于TiO6八面體的中心位置, 而當溫度低于TC時,Γ點的橫光學支振動模軟化, Ti因此偏離中心位移產生位移, 并形成電極化. 而通過X射線吸收精細結構譜(XAFS)在10—15s時間測得的結果顯示[116], Ti總是傾向于與最之靠近的3個O原子成鍵, 從而偏離中心位置, 形成TiO3的配合體,這種情況即使在高于TC的情況下仍舊存在. 但是由于Ti的這種偏離較小, 因而其勢壘較低, 在高溫下可以通過較強熱振動在附近8個靠近O3構型的位置頻繁移動. 這種行為在長程過程下即顯示出Ti原子的平均位置位于晶胞的體心. 第一性原理計算也證實, Ti—O鍵的強共價成分來源于Ti 3d軌道和O 2p軌道的雜化耦合, 是誘發BaTiO3和PbTiO3之間鐵電相的成因[117]. 因此, Ti總是靠近同一平面的3個O原子, 形成TiO3配合體, 在高溫立方相時, Ti在統計上平均地與周圍鄰近的6個O原子形成配位; 而四方相時, Ti則只與鄰近的5個O原子配位; 正交相Ti與4個O原子配位;直到三方相結構, Ti只穩定與其中3個O原子配位, 不再移動.
Atern和 Yacoby[118]對 KTaO3:9%Nb使 用XAFS直接測得的O原子相對于近鄰Nb分布概率顯示更直接的證據. 由于純的KTaO3為先兆鐵電體[119], 高于0 K下顯示為立方順電結構, 而引入Nb可以提高其鐵電相轉變溫度TC. 9%的Nb摻雜的KTaO3顯示的TC為86 K, 但實驗測得結果顯示當溫度達到300 K時, Nb原子在立方相結構下仍然有很大的離子位移, 這說明晶體宏觀畸變的變化并不是通過降低離子位移來實現, 而是通過打亂位移方向來降低畸變程度. 這個結果顯示鈣鈦礦晶格的相變存在很強的有序-無序型機制.
Shuvaeva等[120]通過 XAFS研究了 KNbO3的相變, 發現該晶體的相變同時受位移型和有序-無序型機制的作用. 低溫下的三方相為完全的位移型結構, 而在高溫下其有序-無序型機制則明顯增強. 圖4給出了KNbO3中不同相結構下Nb原子的實際位置, 該示意圖所示模型來自于XAFS測量所得結果[120]. 其中八位模型(eight-site model)對應為理論的有序-無序機制下Nb所在位置,Nb總是沿八個晶向發生位移, 三方相中Nb原子只占據8個中某一位置, 正交相等概率占據沿非極化方向的兩個位置, 四方相占據非極化方向的4個位置, 而順電的立方相中Nb等概率占據所有8個位置. 圖4中顯示, 正交相中Nb的實際位置僅僅略微偏離位移型模型中的位置, 相反在立方相中, Nb原子的實際位置沿a和b方向都大幅偏離位移型模型給出的位置, 更加靠近八位模型的位置. 從圖4可以看出, Nb原子的實際位置與這兩種模型都略有差別, 這說明在Nb原子在局域結構中同時受位移型機制和有序-無序型機制的影響,而在室溫下的正交相中, 這種無序機制相對顯得十分微弱, 難以被探測. 因此, 可以得出結論, 在低溫下KNbO3主要受位移型機制影響, 隨著溫度的上升, 則受有序-無序行為的影響更強.
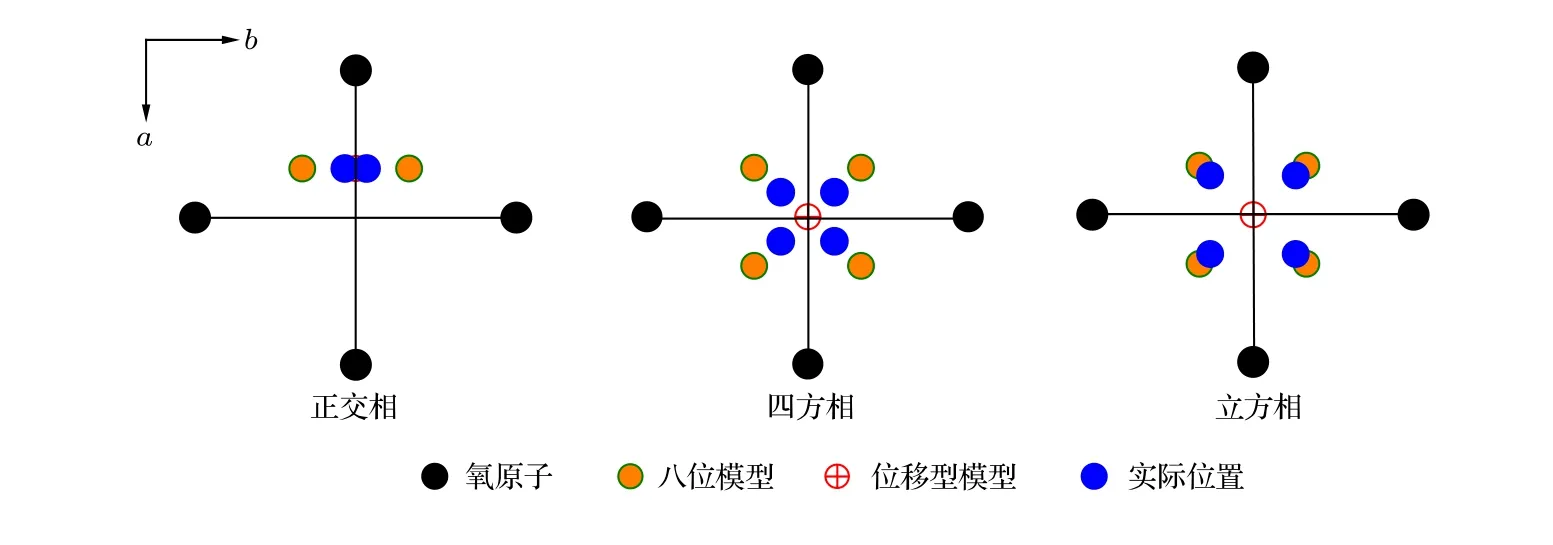
圖 4 根據文獻[120]重畫的KNbO3中Nb原子位置在(001)平面的投影示意圖Fig. 4. Projections of real Nb off-center displacements on the (001) plane redrawn from the Ref. [120].
根據Landau-Ginsburg-Devonshire熱力學唯象理論[121,122], 鐵電相變實際上是中心對稱順電相的自發對稱破缺行為, 產生的鐵電相可以電極化P為序參量去描述系統狀態. Devonshire[121,122]將立方相作為原型, 認為鐵電相僅僅是順電相下的微擾, 因此可以將立方相附近的自由能F以電極化P的形式進行展開, 并用以描述鐵電相的畸變程度. 而因為立方相的中心對稱性,, 因此展開相中不包含奇次項. 在考慮四方相極化強度和施加電場均沿[001]方向的情況下, 其立方相附近的自由能可以寫為
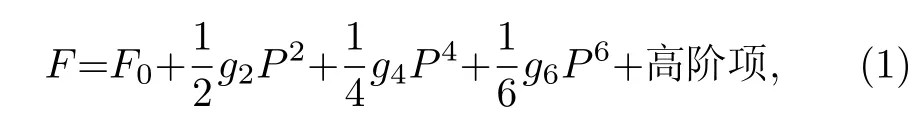
其中F0是P= 0時立方相的自由能;g2,g4和g6稱為Landau系數, 其值與通常溫度有關. 此時,該系統在無外加電場出現極小值的情況為

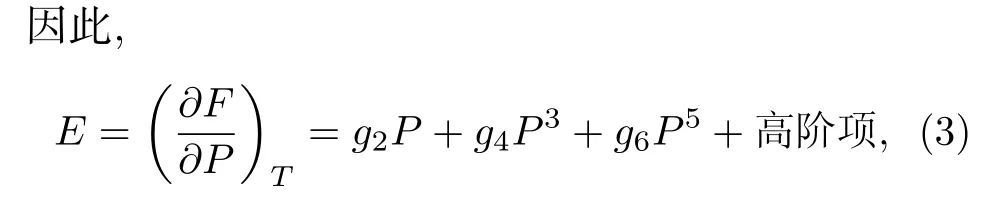
該項等于0時存在兩個解, 一個解是P= 0, 對應于立方的中心對稱相, 另一個解為, 其中P≠ 0, 對應于低于居里溫度時的鐵電相解.

此時,γ是一個正數,T0為小于或等于相變溫度的一個溫度. 若g4為正值, 將高階g6項及以上忽略,此時材料的自發極化強度Ps對應與零場的關系可通過(3)式寫為,
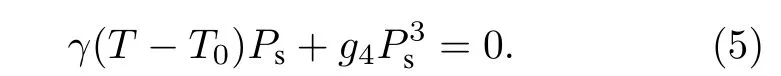
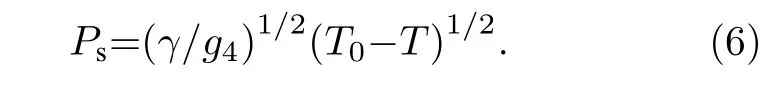
從(6)式可以看出,Ps在居里溫度附近連續地下降為零, 表明該相變為二階相變.
而對于g4為負值的情況, 自由能必須重新包含g6項, 且g6為正值. 因此, 對于E= 0 的平衡條件, 可以寫為
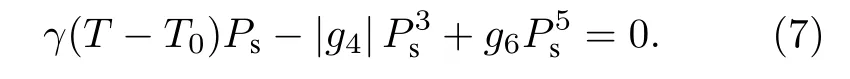
此時即使在等于或略高于相變溫度時, 同樣可以解得兩個值, 一個為Ps= 0, 另一個則是方程
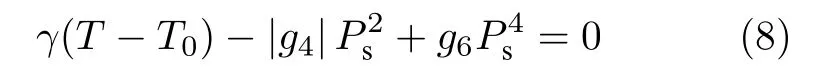
的解, 表明此時順電相和鐵電相的自由能完全相等, 兩相共存,Ps存在等于零和不等于零兩個可以共存的解, 說明該相變為一階相變.
Cochran[123]等通過對 Landau-Ginsburg-Devonshire自由能進行八階展開, 僅考慮二階系數為溫度相關系數, 其余高階系數為常數, 利用KNbO3和K0.5Na0.5NbO3實驗數據計算出總共十個高階項的Landau系數, 成功構建出了K1—xNaxNbO3的自由能函數, 然后對其相圖進行了預測,其預測值與實驗結果十分符合.
4.2 鐵電材料中的壓電行為
壓電材料的壓電系數的表達式為
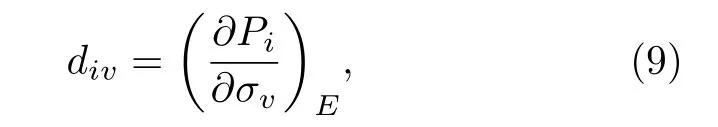
其中d為壓電系數,E為電場強度,P為極化強度,σ為應力強度;i取值范圍為1—3,v取值范圍為1—6. 從(9)式可以看出, 材料的壓電系數實際上是電極化隨應力的梯度, 因此壓電系數實際上表示的是應力作用下電極化的變化幅度. 這種在鐵電材料中的電極化改變實際上可以分為兩類: 一類是應力作用下, 晶格產生形變, 為了使晶格能量處于最低值, 其晶格內部的離子需要重新排列, 從而發生移動, 導致了電極化改變, 這類可以叫做本征壓電效應; 而由于鐵電材料中具有眾多的具有取向的鐵電疇, 并且相互間呈一些特定角度進行排列, 應力作用下, 比如縱向應力σ3, 材料此時沿c方向壓縮,晶格在ab平面拉伸, 導致電疇方向轉向沿ab平面內的方向, 這種轉向的電疇多來自于非180°疇, 其疇壁在應力下相對容易發生移動[124], 從而導致宏觀電極化分量發生改變, 這稱為非本征壓電效應.
鐵電材料的壓電性最顯著的特點是, 在鐵電-鐵電相界處具有極高的壓電系數. 這一現象發生的原因在于, 對于非本征壓電效應來說, 在鐵電-鐵電相界處, 兩個或幾個鐵電相的自由能相等, 比如KNN中的三方-正交-四方相界, 電疇可能沿,和總共26個方向進行取向, 這些極化態之間的能量是相等的, 電疇因而可以很容易地在外場作用下推動疇壁移動來實現轉向, 從而讓極化強度P發生大幅度改變. 對本征壓電效應而言, 多相共存也在實際上降低了不同極化態之間的勢壘, 能量曲線沿各個不同相的極化態之間更加平滑, 因此晶格受同樣外力作用下的離子產生的可逆位移也相對更大, 如圖5所示的正交與三方共存時的能量曲線示意圖[125]. 因此, 鐵電相界的存在可以同時大幅度提升本征與非本征壓電系數, 這也是鈣鈦礦壓電材料的高壓電活性最重要的來源.
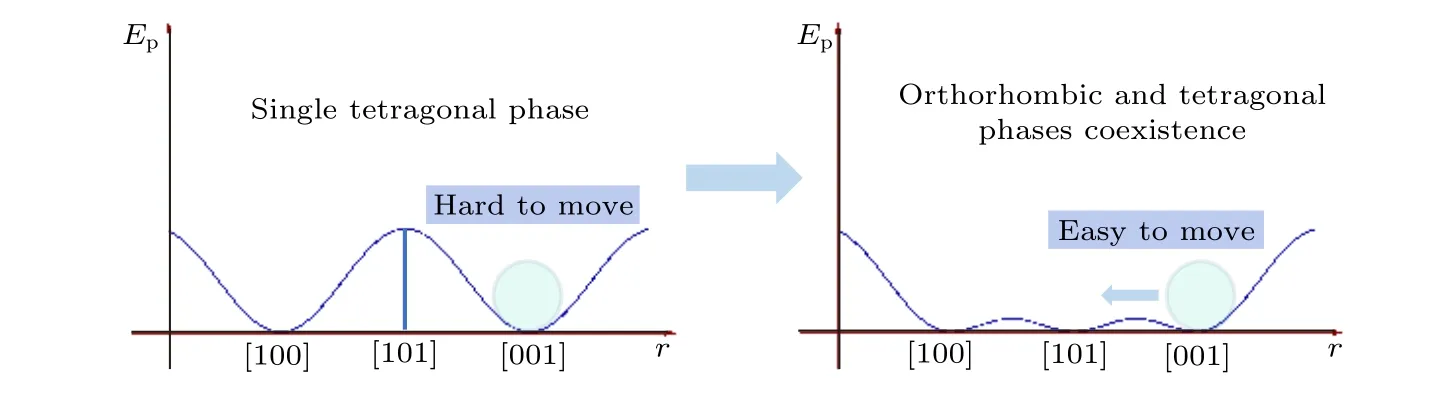
圖 5 B位原子在單四方相與兩相共存時沿[ ]方向的能量分布示意圖[125]Fig. 5. Energy distribution for B-site atom in single tetragonal phase and two-phase coexistence along [ ] direction[125].
4.3 相界構建機制
傳統的Pb基陶瓷中, PZT由三方相的反鐵電體PbZrO3和四方相鐵電體PbTiO3按照一定比例混合構建出準同型相界MPB, 并由此獲得極高的壓電性能. 而在無鉛壓電陶瓷, 比如KNN中,KNbO3和NaNbO3的等量混合并沒有造成大的結構變化, 僅僅是其β角出現一點改變, K含量增加而β角下降[126]. 進一步的研究揭示, 室溫下的KNN在Na含量偏高時是空間群為Pm的單斜結構, 而在K偏高的部分一側為Amm2的正交相結構, 但總體來說兩者在極化方向上差別不大, 用Glazer符號[127]表示兩相分別為和.考慮到KNN中Pm結構的a和c本身差距很小,以致于實驗中都難以被探測到, 而兩者的極化方向都是沿方向, 因此盡管在K/Na = 1附近存在這樣一個相界, 但其對壓電性的貢獻也相當有限, 普遍未摻雜的KNN陶瓷壓電特性都僅在80 pC/N左右.

表 7 處于6配位時的離子半徑表[131]Table 7. The ionic radii in six-fold coordination[131].
目前, 提高KNN基陶瓷壓電性最普遍的做法是通過離子摻雜取代, 提升三方-正交相變溫度或降低正交-四方相變溫度, 誘導在室溫下形成多個鐵電相的共存態. 例如, Li取代A位離子, 誘導正交-四方相變溫度降低至室溫, 形成室溫下的鐵電相界, 該方法可以將KNN陶瓷的d33提升至235 pC/N, 并將TC提升約 470 ℃[128]. 第一性原理研究表明, Li摻雜可能大幅提高了A位離子p軌道與O 2p的軌道雜化[129]. 這與PbTiO3的形成原因類似, Pb與O的軌道雜化誘導了四方相的形成, 并提高極化強度, 增加TC. 但這仍有疑問,由于以上研究均是結合虛晶近似(virtual crystal approximation, VCA)進行的, 難以區分到底是Li, Na和K中的具體哪一種元素造成的A位離子與O的強雜化, 且Li的電子軌道中不包含p軌道.更關鍵的是無論Li, Na還是K的電負性都明顯低于Pb, 最高的Li也僅僅與Ca的電負性類似, 但CaTiO3中A位不存在強共價作用, 且其是典型的立方相結構, 因此關于Li摻雜KNN誘導四方相的起源尚有待進一步研究.
從B位離子取代的角度來看, 通常來說, 認為大尺寸的B位可以給O6八面體內部更高的化學壓力, 從而驅動晶體結構向三方相轉變. 這在PbTiO3的計算中也有所證實, 四方相的PbTiO3可以在高靜水壓下轉變為三方相結構[130]. 因此,Zr和Hf等容易驅動KNN向三方相轉變, 并且由于摻入的B位離子半徑較大, 其在氧八面體內的離子位移受限降低, 因而正交-四方轉變溫度和居里溫度隨之降低. 半徑與Nb相似的Ta則對KNN的相變溫度影響十分微弱, 平均每0.01 mol降低正交-四方轉變溫度TO-T約4 ℃, 提升三方-正交相變溫度TR-O約2.3 ℃[8]. 值得注意的是, Sb的取代對提升KNN材料的壓電性質十分明顯, Sb可以迅速地提高TR-O, 同時降低TO-T, 由于這個特性, Sb已經成為KNN基材料摻雜過程中最重要的取代元素, 絕大部分高壓電的KNN材料都含有Sb. 實際上Sb的取代行為與Zr和Hf十分類似,但是由于其名義上的化合價為+5價, 與Nb相同,因而其不容易破壞晶格的長程有序性, 相對容易進入晶格. 而Zr和Hf的化合價都是+4價, 研究顯示過量的Zr很容易聚集在晶界處, 影響晶粒生長,因此 Zr和 Hf的取代通常都伴隨著 Ba2+, Sr2+,Ca2+或者 (Bi0.5Na0.5)2+等+2 價離子. 但是, 根據Shanon[131]給出的離子半徑 (見表 7), Sb5+的離子半徑比Nb5+要更小, 在KNN中取代時不會對O6八面體提供內生的化學壓力, 導致TR-O上升和TO-T下降. 本課題組研究Ga2O3摻雜對KNN的影響時發現[132], 小半徑的Ga3+進入KNN晶格B位反而會提升TO-T, 與大尺寸離子對相界的作用基本相反. 在考慮到Sb在高溫下容易被還原為三價離子, 懷疑KNN陶瓷中可能是+3價態的Sb在推動KNN相變溫度的改變, 這需要進一步的研究去證實. 總之, 根據以上提及的摻雜機制, 通過調整不同摻雜物的比例, 控制各個鐵電相之間的相變溫度, 就可以在KNN基材料中設計出室溫下三方-正交共存、正交-四方共存甚至三方-四方共存的鐵電相界, 從而獲取高壓電性的無鉛材料.
另外, 需要注意的是, 不同價態的離子在摻雜過程中容易造成晶格點缺陷, 常見的如CuO摻雜,六配位的Cu2+離子半徑為0.73 ?, 容易進入晶格B位, 此時的化學反應為
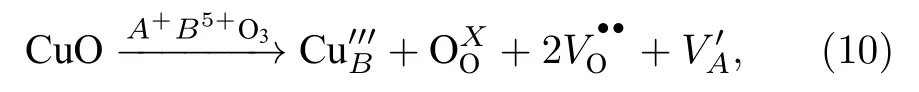
4.4 第一性原理計算
第一性原理計算基于密度泛函理論(DFT),DFT理論將晶格內部的多電子薛定諤方程轉化成為一個求解電子氣密度函數, 在形式上近似于單電子薛定諤方程的Kohn-Sham方程來求解體系基態能量. 然后通過靜態晶格來實現對材料力學、聲子頻率、有效電荷、介電常數、壓電常數、彈性常數和極化強度等宏觀性質的計算. 在這個過程中, 由于第一性原理計算本質上是直接求解材料本身的多電子薛定諤方程, 因此不需要提供任何實驗收集的經驗參數. 目前許多對鈣鈦礦壓電材料的重要機理解釋都來自于第一性原理計算, 比如Ti—O鍵的軌道雜化, 這直接證明了鐵電性起源于長程庫侖力之間的競爭, 并導致了離子位移; 相反, 短程排斥力則容易促使形成高對稱的順電相.
第一性原理計算在Pb基材料之中研究相對廣泛, 比如證明了弛豫鐵電體中的高壓電應變來自于極化方向的偏轉[134], 預測了高壓下純PbTiO3存在一個MPB相界, 并存在高壓電活性等[130]. 而在無鉛壓電材料中, 相關研究仍然相對較少, 尤其是近些年熱門的KNN材料. 在此, 本文對第一性原理計算在KNN中的研究進行簡要介紹.
長久以來, Ta通常被用作KNN壓電材料的摻雜取代元素之一, 并通過和Li, Sb等元素共同摻雜誘導室溫下形成鐵電相界, 獲得了具有高壓電活性的KNN基陶瓷, 但Ta取代Nb占據B位是如何對晶格產生影響的卻缺少研究. Suewattana和Singh[135]使用第一性原理計算研究了純的KNN合金與Ta摻雜的KNN (KNNT)合金的局域結構和動力學. 研究發現Ta取代后的局域結構具有比Nb更小的離子位移. 盡管Nb與O的距離相對Ta更近, 但Nb的力常數反而比Ta更小. 另外, 通過獲得的諧性徑向分布函數(PDF)發現, 純的KNN和KNNT在高溫下的第3個分布峰即Nb-Nb或Ta-Ta的分布峰仍然保持尖銳, 說明Nb和Ta的分布仍然保持相對有序的結構, 但KNNT中Ta全部的峰形都相對Nb更加尖銳, 顯示出Ta具有更大的力常數, 這被認為是導致實驗中出現Ta摻雜后居里溫度降低和介電常數增加的主要原因.
Voas等[136]使用第一性原理結合準隨機結構(SQS)研究了 K0.5Na0.5NbO3在R3c和Pm結構下的A位離子分布. 通過與中子衍射測試的數據進行對比, SQS能夠準確給出鈣鈦礦結構中A位離子的局域分布形式. 通過結構計算顯示, Na更傾向于向靠近K遠離Na的方向產生位移, 而又同時盡可能與整體極化方向保持一致. 而在基態相R3c和室溫相Pm之間, Na-O的交互作用變化最為明顯, 這顯示Na-O的交互作用是導致從R3c到Pm相變的驅動力.
室溫下的KNN陶瓷具有比KNbO3(KN)更高的壓電系數[137], 但兩者都為正交相結構, 鐵電相變在室溫下未對兩者的壓電性能產生貢獻. 本課題組通過第一性原理計算結合取向平均的方法[138,139], 直接比較計算了正交相 KN 和 KNN 的單晶與陶瓷的壓電性能, 研究發現KNN陶瓷明顯具有比KN更高的壓電性能, 其d33相較KN增加了約70%, 這是與實驗觀察所得一致, 計算結果顯示兩者正交相陶瓷的d33都主要來源于單晶中d15和d33的貢獻. 進一步研究發現, KNN與KN的Born有效電荷相差不大, 說明電子部分的貢獻兩者幾乎沒有差別. 在該研究中, KNN中的K和Na按照沿鈣鈦礦方向交替進行排列, Na較小的半徑會引起自身相對大的離子位移, 在KNN中產生增強的鐵電性; 同時, Na會廣泛引起自身及鄰近的O產生明顯增強的離子位移隨應變的響應, 如表 8 所示, 沿a方向靠近 Na 的 OI,2在應變η5下 的 響 應 是 靠 近 K的 OI,2的 幾 乎 五 倍 ,KNN中Na自身的響應更是K的九倍. 因此, 正是引入較小半徑的Na導致的Na自身和鄰近O的移動空間增加, 進而增強了應變作用下的離子位移響應, 導致室溫下的KNN比KN具有更高壓電性.
Yang等[129]則利用DFT加上虛擬晶胞近似(VCA)方法調查了Li摻雜的KNN材料, 對比了其電子態密度分布及壓電應力常數e33, 驗證了Li摻雜后增強的壓電性能. 其結果顯示Li摻雜增強了O 2p軌道和Nb 4d軌道的雜化, 減小了Nb-O鍵距離并增強其畸變程度. 這些結果被認為是Li摻雜后壓電性能增強的主要原因.
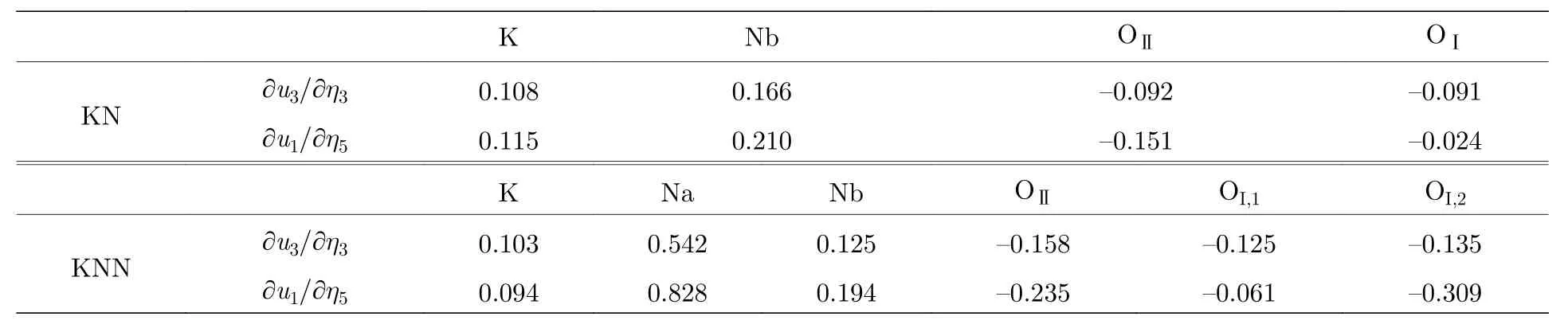
表 8 不同結構下原子內坐標隨應變的梯度, 注意OI位于Bmm2不包含Nb原子的(010)平面, KNN中OI,1和OI,2沿a方向分別靠近K和Na原子[138]Table 8. Internal atomic coordinate gradients as a function of strains in different structure, noted that OI is located at the(010) plane without Nb atoms in Bmm2, OI,1 and OI,2 are close to K and Na along a axis, respectively[139].
Li等[140]對CaZrO3摻雜的KNN的電子結構進行了計算. 研究發現Ca在A位相對更容易取代Na, 并且Ca摻雜會導致KNN的費米面向高能量的導帶移動, 從而降低帶隙; 而Zr的摻雜則會讓費米面向價帶移動, 提高帶隙寬度. 此外, Ca取代A位具有更大的電負性, 從而誘導KNN沿c方向拉伸, 因此KNN逐漸從O相轉變為T相, 這是導致CaZrO3取代讓KNN具有更高性能的主要原因. 關于K/Na = 1附近KNN的相變問題一直爭論已久, Liu等[141]研究K1—xNaxNbO3體系的相變,發現隨Na組分的增加, 該體系可能經歷O-MO的相變過程, 顯示出該類材料在0.3 <x< 0.8所具有的并不是嚴格意義上的正交相O, 反而是一個低對稱性的單斜相M, 并推測該單斜相可在常規R, O和T三相中扮演中間相的角色, 讓各個極化態之間相互轉向更容易, 從而提高材料的壓電活性. 哈爾濱工業大學的Li等[142]利用第一性原理計算結合VCA的方法詳細研究了K1—xNaxNbO3體系的壓電系數, 并預測其MPB出現在x=0.52附近. 該研究同時計算了體系的結構參數、體積模量和禁帶寬度, 并指出極化強度在x= 0.5時從[011]到[001]轉向具有比純KNbO3更低的能量壁壘, 這是導致其具有增強壓電的主要原因.Yang等[143]使用第一性原理計算了不同Na濃度下的KNN結構與體系總能, 發現當Na濃度逐漸增加時, 出現一個O-T相轉變, 并導致了增強的壓電特性, 該結果證明了高d33來源于相變. 計算結果還表明NbO6八面體的形變與材料整體的相變起源于氧八面體內Nb—O鍵長度的變化. 此外, 在計算中, KNN在Na含量為0.55時出現一個最高的壓電常數e33= 6.77 C/m2, 而通過與實驗進行對比, 實驗中陶瓷的d33也在該組分展現了最高的壓電性d33= 203 pC/N, 與計算結果的趨勢一致,Yang等[143]認為KNN陶瓷中壓電性能的這些變化可能來源于組分波動與相結構的轉變.
5 結論與展望
通過添加一定的離子或者化合物調節KNN基無鉛壓電陶瓷的TR-O和TO-T相變溫度至室溫附近, 構建室溫附近的新型相界, 是獲得高性能的KNN壓電陶瓷最為有效的手段之一. 通過織構化工藝, 可以有效地提升KNN基壓電陶瓷的性能,是未來最為值得關注的制備方法之一. 近年來, 國內外研究者們針對不斷提升KNN基無鉛壓電陶瓷的壓電性能, 取得了較大的突破, 但是KNN基壓電陶瓷的綜合性能與鉛基壓電陶瓷相比仍存在一定的差距, 因此, 還需要在組分設計、工藝優化等方向繼續探索.
對于KNN基無鉛壓電陶瓷, 壓電性能的提升會伴隨著陶瓷體系TC的下降; 當KNN陶瓷在小于居里溫度的溫度范圍內存在另一個多晶型相變時會導致陶瓷熱穩定性下降. 這是KNN基陶瓷與PZT陶瓷相比最大的不足之處, 這將影響KNN陶瓷的實際應用. 如何獲得同時具有高壓電性與高溫度穩定性的KNN基無鉛壓電陶瓷仍然是未來研究的難點與重點之一.
另外, KNN基無鉛壓電陶瓷因為性能對成分的敏感性, 燒結溫區較窄以及工藝重復性差, 不利于KNN基無鉛壓電陶瓷的大規模生產. 以實際應用為前提, 如何解決KNN基無鉛壓電陶瓷的組分、溫度敏感性是關系到該體系規模化應用的關鍵.
而在理論方面, 一方面KNN中K/Na的分布結構仍值得進一步研究, 因為盡管K和Na的價電荷較低, 離子位移對極化強度貢獻相對較Nb和O更小, 但K/Na的分布結構對壓電性也具有一定影響[138,139], 尤其是對鄰近的 O 影響最為明顯. 另一方面, 有限溫度下的壓電活性在KNN中仍然值得探討, 關于該方面的論述可能可以從理論上對PPB相界的低熱穩定性進行解釋. 此外, 離子摻雜、多組元復合對晶格的作用需要深入研究, 尤其是 Li和 Sb 的摻雜, CaZrO3, Bi(Zr, Hf)O3與 KNN的復合, 以便更好地理解相變的驅動力來源和高壓電活性的起源.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
