Rett綜合征致病基因突變與臨床表型的研究
2020-07-17 11:30:04王寶田唐久來
安徽醫學 2020年6期
楊 李 王寶田 唐久來 吳 德
Rett綜合征是一種主要累及女性患兒,嚴重影響運動及智力發育的遺傳性罕見病,女童患病率為1/15 000~1/10 000,男性患兒更為罕見,約95%的患兒由位于X染色體長臂上的甲基CpG結合蛋白2(methyl CpG binding protein 2,MECP2)基因突變所導致,其他如細胞周期蛋白依賴激酶5(cyclin-dependent kinase like 5,CDKL5)、叉頭盒蛋白G1(forkhead box protein G1,FOXG1)等基因突變也可導致本病的發生[1]。典型Rett綜合征經過早期的正常發育至6~18個月齡后,出現已獲得技能的迅速退化、典型手部運動、小頭畸形、自閉癥、共濟失調以及呼吸節律紊亂等;除了典型Rett綜合征外,還有非典型的變異型,如先天性變異型、恢復部分言語能力的語言保留型及早發癲癇型[1-2]。本研究總結Rett綜合征臨床表現、分析突變基因,擴展臨床表型,提高對本病的早期診斷。
1 資料與方法
1.1 一般資料 本研究選取2017年1月至2019年12月在安徽醫科大學第一附屬醫院兒科就診,明確診斷為Rett綜合征的患兒,收集臨床資料,病例采集前患兒監護人均已簽署知情同意書。
病例1:女性,診斷年齡3歲7個月,系第1胎第1產,患兒1歲余可獨走,但僅能無意識發“ba、ma”音,曾以“孤獨癥”外院治療,約2歲半時出現顯著倒退,表現為獨走不穩,出現雙手反復交替用手指觸碰舌頭后放下的手部刻板動作,不再主動抓物,僅會發“a”音,不與人交流。3歲6個月出現癲癇,全面性強直發作,現接受抗癲癇藥物與生酮飲食治療。父母表型正常,否認家族史。
病例2:女性,診斷年齡1歲3個月,系第2胎第2產, 6~13個月期間身高增長緩慢,僅2 cm,現不會爬行,可扶站,無意識發“ba、ma”音,手部精細動作差,伴有刻板動作搓手、磨牙,頭圍43 cm,睡眠質量較差、節律紊亂,易激惹。患兒姐姐2歲8個月表型正常,父母表型正常,否認家族史。
病例3:女性,診斷年齡2歲,系第1胎第1產,6月齡前發育基本正常,1歲2個月時家長發現患兒只會無意識發“yi、ya”音,雙手出現無法穩定抓握,1歲5個月出現手部刻板動作,喜搓手、用手拍頭,與人交流欠佳,現患兒2歲,偶喜尖叫,與人無眼神交流,多動,手部刻板動作無改善,獨走欠穩。父母表型正常,否認家族史。
病例4:女性,診斷年齡4歲,系第2胎第1產,發育里程碑稍落后,2歲余可獨走,但姿勢異常,可有意識發“ye、ma”音,其后逐漸出現運動認知倒退,手拍口唇、搓手等刻板動作,眼神呆滯,情緒易激動,4歲時癲癇頻發,為全面性強直陣攣發作和復雜部分性發作。我院檢查示:高磷血癥、腎源性血尿;現接受抗癲癇藥物及生酮飲食治療。父母表型正常,否認家族史。
病例5:男性,診斷年齡3歲1個月,系第1胎第1產,10月齡獨坐不穩,無意識發“ba、ma”音,伴有通貫手,曾間斷隨訪于我科,1歲7個月獲獨坐能力,3歲1個月時已無法獨坐,不會發“ba、ma”音,四肢肌力、肌張力低下,出現脊柱側彎,睡眠節律紊亂,喜磨牙。患兒母親智力低下,無法生活自理,父親邊緣智力,基本生活與體力勞動無障礙。
1.2 方法
1.2.1 標本采集 采集患兒及父母全血2 mL,提取DNA并質檢,合格后進入分析步驟。
1.2.2 測序與生物信息學分析 構建全外顯子文庫,通過illumina公司NovaSeq 6000系列測序儀進行高通量測序(PE150),測序覆蓋度不低于99%。使用Burrows-Wheeler Aligner(BWA)等軟件篩選及過濾,獲得可靠高質量突變數據。對檢測到的突變數據進行各大數據庫,如單核苷酸多態性數據庫(the Single Nucleotide Polymorphism Database,dbSNP)、千人基因組(1000 Genomes Project)、外顯子組整合數據庫(the exome aggregation consortium,ExAC)、外顯子測序計劃(Exome Sequencing Project,ESP)等頻率數據庫,在線人類孟德爾遺傳數據庫(Online Mendelian Inheritance in Man,OMIM)、人類基因突變數據庫(Human Gene Mutation Database,HGMD)、臨床相關的序列變異(Clinically Relevant Sequence Variations, ClinVar)等的關聯注釋。借助Provean、Polyphen2-HVAR、SIFT、Revel、M-Cap等蛋白結構預測軟件進行危害性分析,篩選可能影響蛋白結構的有害變異。
1.2.3 Sanger法一代測序驗證 根據需要驗證的位點進行引物設計,PCR技術擴增,對患兒及其父母樣本測序驗證。
1.2.4 危害性評級 根據注釋信息、頻率數據庫等依據美國遺傳學會(American College of Medical Genetics,ACMG)2015診斷指南,綜合評定危害等級為5級:致病、可能致病、良性、可能良性及意義未明。
2 結果
2.1 臨床特點 病例1~4均符合典型Rett綜合征的臨床診斷標準,病例5符合非典型Rett綜合征臨床診斷標準[2],見表1。病例1除了典型Rett綜合征的表現外,同時伴發有癲癇;病例2頭圍43 cm,低于正常同齡兒童2個標準差;病例4伴有癲癇、堿性磷酸酶升高、高磷血癥、腎源性血尿;病例5為男性患兒,雖然該病例1歲2個月即明確存在致病突變,但至3歲后癥狀趨于明顯。

續表1
2.2 分子遺傳學檢測結果
病例1:檢測到一個位于X染色體的雜合基因突變 MECP2[c.808(exon4)C>T,p.Arg270Stop,217],即第808號核苷酸由C突變為T,導致翻譯產物的第270號的精氨酸以后的217個氨基酸無法翻譯。Clinvar數據庫和HGMD數據庫中已確定相同的致病變異或者有相同的氨基酸改變。患兒父母上述位點均未檢測到變異,故屬新生變異,綜合評定危害等級:致病。Sanger測序驗證見圖1-A。
病例2:檢測到一個位于X染色體的雜合基因突變MECP2[c.763(exon4)C>T,p.Arg255Stop,232],即第763號核苷酸由C突變為T,導致翻譯產物的第255號的精氨酸以后的232個氨基酸無法翻譯。Clinvar數據庫和HGMD數據庫中已確定相同的致病變異或者有相同的氨基酸改變。患兒父母上述位點均未檢測到變異,故屬新生變異,綜合評定危害等級:致病。Sanger測序驗證見圖1-B。
病例3:檢測到一個位于X染色體的雜合基因突變MECP2[c.799(exon3)C>T,p.Arg267Stop,220],即第763號核苷酸由C突變為T,導致翻譯產物第267號的精氨酸以后的220個氨基酸無法翻譯。Clinvar數據庫和HGMD數據庫中已確定相同的致病變異或者有相同的氨基酸改變,PubMed查詢到該變異致病性的文獻報道。患兒父母上述位點均未檢測到變異,故屬新生變異,綜合評定危害等級:致病。Sanger測序驗證見圖1-C。
病例4:檢測到一個位于X染色體的雜合基因突變MECP2[c.916(exon4)C>T,p.Arg306Cys],即第916號核苷酸由C突變為T,導致翻譯產物第306號氨基酸的精氨酸翻譯成半胱氨酸。Clinvar數據庫和HGMD數據庫中已確定相同的致病變異或者有相同的氨基酸改變。患兒父母上述位點均未檢測到變異,故屬新生變異,綜合評定危害等級:致病。Sanger測序驗證見圖1-D。
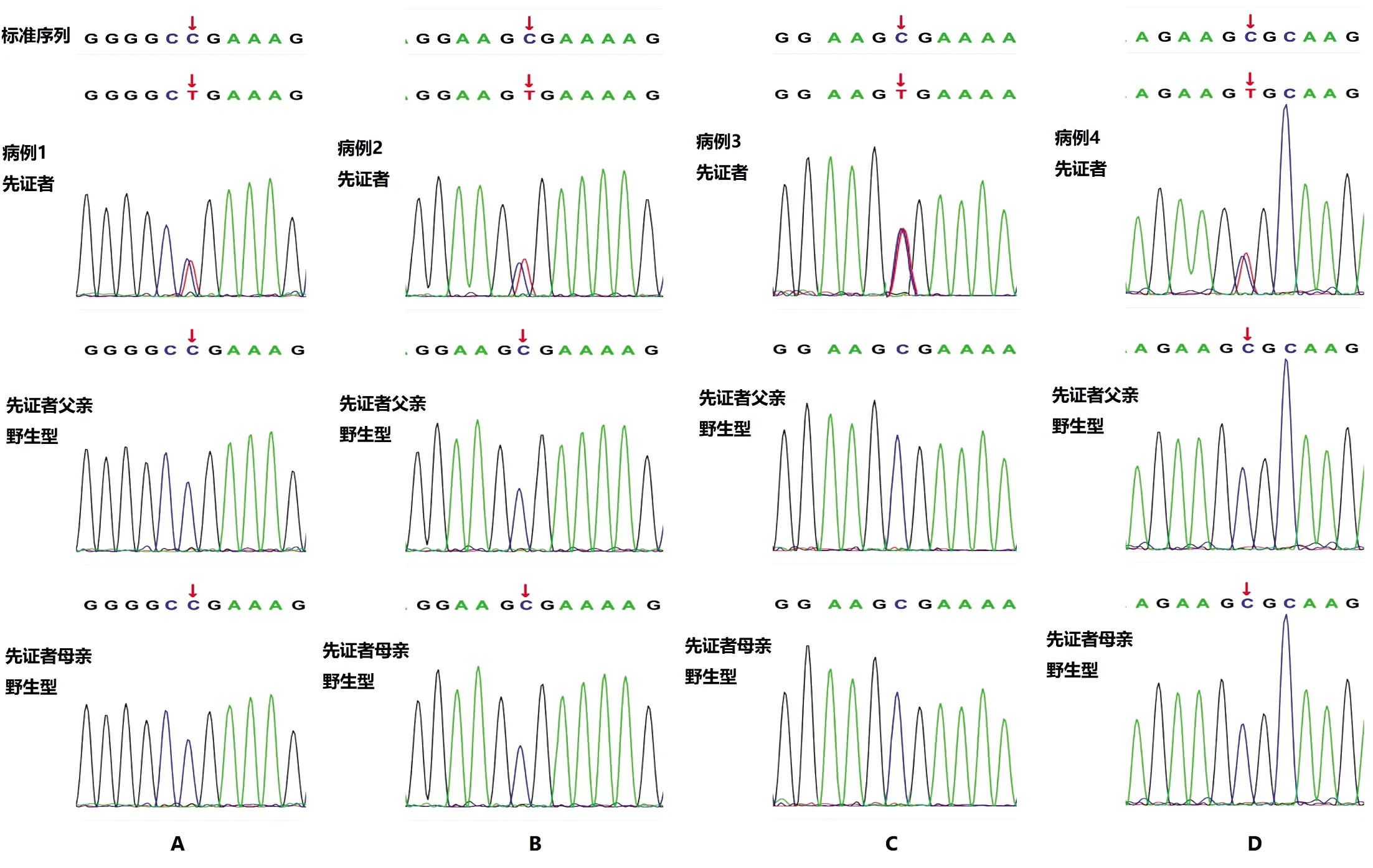
圖1 突變Sanger測序驗證結果
病例5:檢測到一個位于X染色體的半合子基因突變MECP2[c.1148(exon4)_c.1198(exon4)del,insC,p.Leu383Profs*5]從編碼序列1 148位置到1 198位置缺失50個堿基,導致其翻譯產物從第383位異亮氨酸開始,再翻譯5個氨基酸后終止;經蛋白結構預測軟件(Provean、Polyphen2-HVAR、Polyphen2-HDIV、M-Cap、Revel、Mutationtaster)2種以上統計方法預測該變異對基因及產物有影響。在正常人數據庫(dbSNP、千人南方、千人基因、ExAC等)中未見收錄,OMIM、HGMD、Clinvar數據庫中未發現疾病相關性報道。患兒父親為野生型,母親為雜合,故該患兒致病突變來源于其母親,綜合評定危害等級:致病。Sanger測序驗證見圖2。
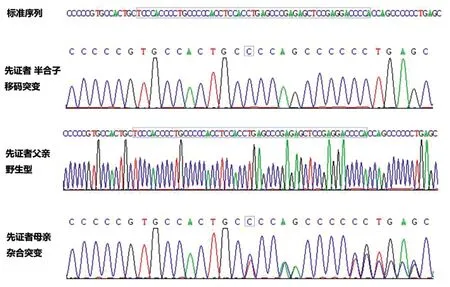
圖2 突變Sanger測序驗證結果
3 討論
Rett綜合征是一種以運動、語言發育退化,以及典型的手部刻板動作等為特征的遺傳性疾病,上世紀60年代奧地利醫生Rett首次報道[3]。Rett綜合征患兒早期生長發育史大致正常,但隨后出現非連續性的病程進展,包括:停滯期、快速退化期、平臺期及后期運動退化期4個階段。在快速退化期,患兒會部分或者全部喪失有目的的手部功能和語言能力,同時出現運動障礙、呼吸異常,自閉特征逐漸顯現,以及癲癇發作等[2,4]。
Rett綜合征公認并且最新的診斷標準是(國際)Rett研究聯盟(Rett Search Consortium)于2002年診斷標準基礎上2010年再行修訂的版本,標準中指出:臨床診斷典型Rett綜合征需滿足全部的主要標準和排除標準,雖然支持標準在典型Rett綜合征中較為常見,但不是診斷的必要條件;而非典型Rett的診斷確立需至少滿足2條主要標準,且同時達到5條支持標準。由于標準中所羅列的臨床特征多具有年齡依賴性,即臨床表現到一定年齡階段才會顯現出來[2],低年齡兒童的診斷的難度要大于年長兒。因此,在實際臨床工作中基因學檢測有助早期診斷,本研究病例2、病例5是基于基因學的早期診斷,其中病例5早期診斷 “可能非典型Rett綜合征”,后期隨訪中其臨床癥狀趨于明顯,診斷為“不典型Rett綜合征”。
癲癇雖未列于診斷標準中,卻是Rett綜合征常見的臨床表型。60%~80%的Rett綜合征患兒合并癲癇,在非典型Rett綜合征和嚴重表型的Rett綜合征患兒中發病率更高,常見的發作類型為復雜部分性以及全面性強直發作[5]。本研究中,病例1為全面性強直發作,病例4為復雜部分性以及全面性強直-陣攣發作。需要指出的是,Rett綜合征的呼吸節律紊亂可導致呼吸暫停、發紺、凝視,以及肌張力障礙等類似癲癇癥狀。本研究病例4同時伴有堿性磷酸酶升高、高磷血癥、腎源性血尿,但該患兒未見有其他未明意義突變關聯表型,且既往文獻中未見描述,本研究為首次報道該表型。
Rett綜合征絕大多數是由位于X染色體長臂上MECP2基因突變所致,約5%的患兒攜帶突變基因CDLK5、FOXG1或癲癇或智力障礙相關的突變基因。本研究中病例均為MECP2基因突變所致,其中病例1、2、3、4在Clinvar數據庫和HGMD數據庫中已確定相同的致病變異,或者有同樣的氨基酸改變[6-7];病例5為較為罕見的男性患兒,其突變遺傳于有表型母親,尚無相同突變的文獻報道。國內報道1例MECP2基因突變致Rett綜合征男童于18月齡時因“呼吸衰竭”死亡[8]。由于MECP2基因位于X染色體上,其突變被認為對發育中的男性胎兒及出生后男嬰是致命的,故Rett綜合征的男性數量較少,國內一項較大樣本Rett綜合征回顧性研究對象也均是女性患兒[7]。
MECP2編碼甲基-CPG結合蛋白2,為核蛋白的一種,在每個組織細胞中均有表達,在神經元中尤其豐富,主要負責翻譯后的修飾作用。MECP2作為一個轉錄調控因子特異性地與甲基化DNA結合,募集蛋白伴侶和調節復合物來修飾轉錄活性。MECP2有3個主要的功能域:甲基CpG結合域(methyl-CpG-binding domain,MBD)、轉錄抑制域(transcription repression domain,TRD)、C終端域( C-terminal domain,CTD)。本研究中,病例1、2、3、4突變位于TRD區域,可導致該區域蛋白結構的改變,影響與甲基化的 DNA結合,進而影響MECP2轉錄抑制功能;病例5的突變位于CTD區域,突變可所致的結構改變,影響介導染色質折疊能力(見圖3),參考文獻[9]繪制。除了突變位點外,突變類型也是影響表型的重要因素,MECP2基因突變致所編碼蛋白氨基酸截短突變的臨床表型最為嚴重,而大多數的末端截短突變與錯義突變的臨床表型則較為溫和[9]。本研究中,3例患兒為氨基酸的截短突變,1例移碼突變,1例患兒為錯義突變,但是錯義突變病例4的表型仍較嚴重。由于Rett綜合征雜合女性患兒是正常和突變MECP2的嵌合體攜帶者,因此臨床表型的差異也與X染色體的差異性失活有關。同樣,修飾基因的突變也可能減輕或增強患者的臨床癥狀[10],這可能是本例錯義突變患兒表型仍較嚴重的原因。
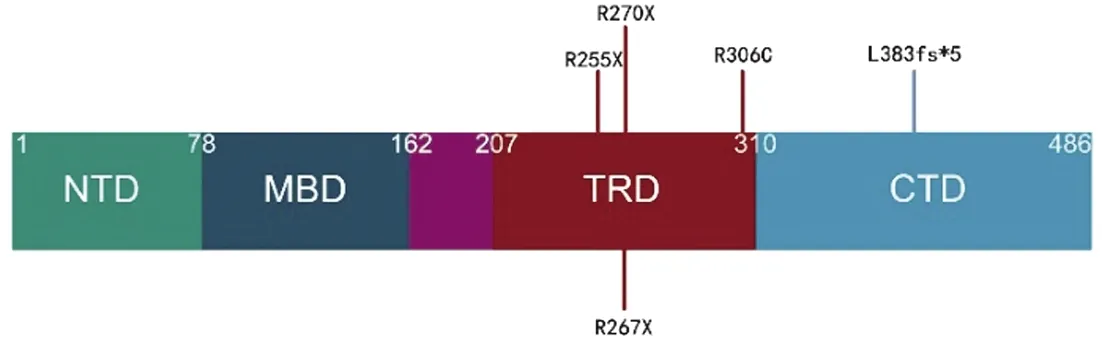
圖3 MECP2突變位點示意圖
綜上,Rett綜合征是一種累及多器官系統的遺傳性疾病,目前尚無特異性有效治療手段,一些藥物仍然處在臨床前研究階段[11],需要個體化的綜合康復治療,其中生酮飲食對癥狀有改善作用,尤其是合并癲癇的Rett患兒[11-12]。結合其遺傳學病因,目前有2種治療研究思路,一種直接針對MECP2突變,對MECP2下游通路的靶點進行治療;另一個潛在途徑是基因療法,即將正常的MECP2導入細胞進行治療[4,9,13]。最近,一個靶向基因治療罕見遺傳性眼病Leber先天性黑矇在美國被批準應用,這可能也是未來治療Rett綜合征的重要方法[14]。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國民間療法(2021年5期)2021-06-09 09:21:04
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
飲食科學(2017年5期)2017-05-20 17:11:53
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
西南軍醫(2015年4期)2015-01-23 01:19:30
