
免疫性血小板減少癥轉(zhuǎn)化為RUNX1基因陰性的兒童急性巨核細胞白血病臨床分析
2020-07-17 11:30:06陳天平王亞萍屈麗君
安徽醫(yī)學 2020年6期
陳天平 王亞萍 程 杰 許 喆 李 艷 汪 儉 屈麗君
免疫性血小板減少癥(immunothrombocytopenia, ITP)是由體液和細胞免疫介導骨髓巨核細胞成熟障礙所致血小板生成不足,和/或免疫因素所致血小板過度破壞的一類自身免疫性疾病,其發(fā)病機制復雜多樣,具有明顯的異質(zhì)性[1]。近年來,隨著分子遺傳學技術(shù)的發(fā)展,該病的發(fā)病機制得以進一步闡述,部分既往診斷為慢性ITP的患兒最終確診為遺傳性血小板減少癥。其中,RUNX1基因突變的具有急性髓系白血病(acute myeloid leukemia,AML)傾向的家族性血小板疾病(familial platelet disorder with predisposition to acute myeloid leukemia, FPD/AML)具有易于向AML轉(zhuǎn)變的特征[2-3]。本研究通過對2例ITP轉(zhuǎn)化為RUNX1基因陰性的急性巨核細胞白血病(acute megakaryocytic leukemia,AMKL)患兒進行臨床及實驗室檢查,并結(jié)合國內(nèi)外相關(guān)文獻,探討此類罕見疾病的臨床特征。
1 資料與方法
1.1 一般資料 收集安徽省兒童醫(yī)院血液科2018年1~12月收治的AMKL患兒2例,均為女嬰,就診時年齡較小,都因出血癥狀就診。追問病史,既往皆有ITP病史,曾行骨髓細胞學檢查排除血液造血系統(tǒng)疾病,并行自身抗體譜系等檢查排除自身免疫性疾病。所有患兒均否認陽性血液系統(tǒng)疾病家族史,亦否認異常孕產(chǎn)史。患兒一般資料見表1。
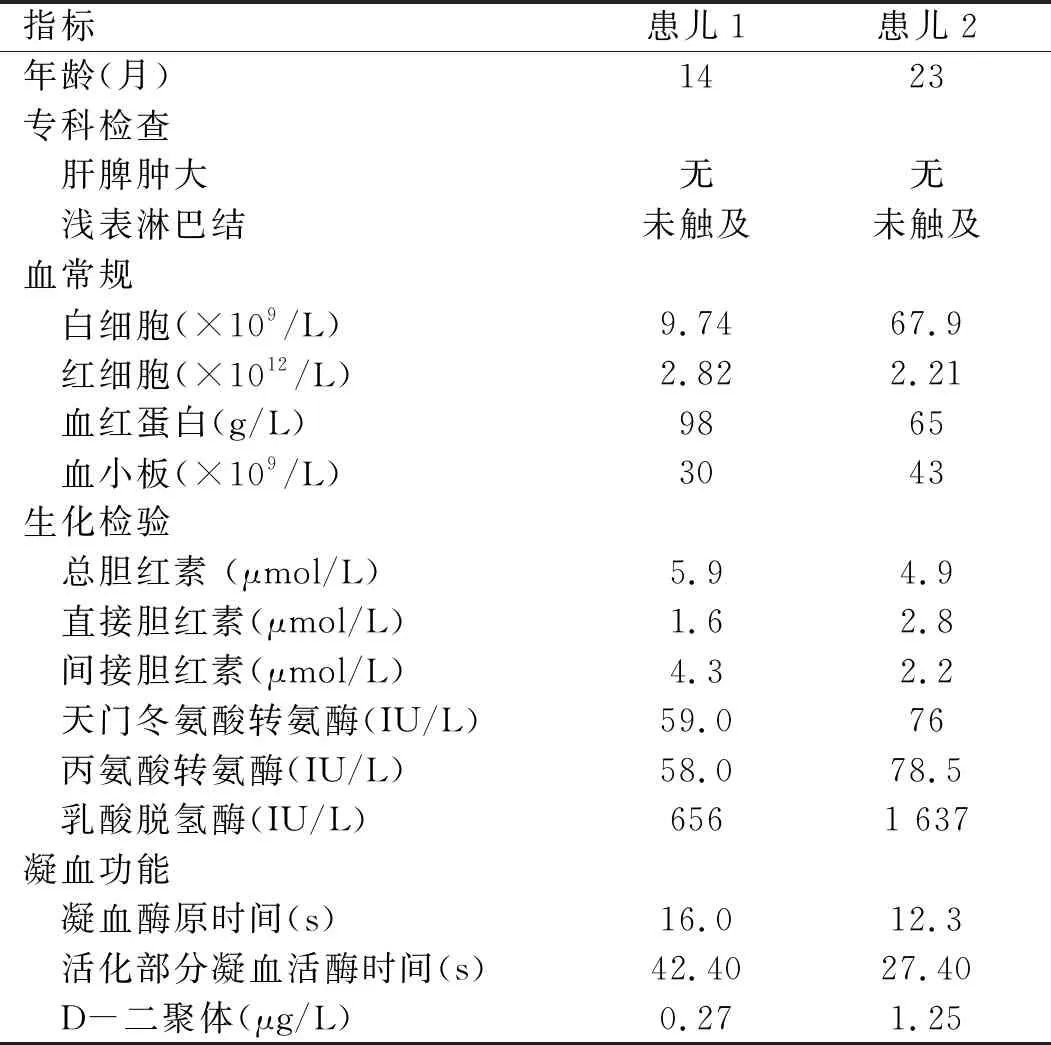
表1 2例患兒一般資料
1.2 檢查方法
1.2.1 骨髓細胞檢查 采用局部麻醉,選取髂后上棘為穿刺部位,將抽取的骨髓液迅速涂片3~5張,實行瑞-吉姆薩染色,使用低倍鏡對骨髓增生度進行觀察判斷,油鏡下觀察骨髓細胞的分類、形態(tài)及計數(shù)。
1.2.2 骨髓活組織檢查 局麻處理后,取髂后上棘行骨髓活檢穿刺,鉆取0.4~1.0 cm的骨髓組織。使用Bouin液對骨髓組織固定,經(jīng)脫水、浸透處理后運用塑料包埋,將其制成切片,行HE染色和免疫組化染色。采用顯微鏡對骨髓病理形態(tài)進行觀察。
1.2.3 流式細胞學分析 取骨髓液1~2 mL,EDTA抗凝,調(diào)整細胞數(shù)至(4~10)×109/L,取100 μL骨髓液加入單克隆抗體20 μL,分析60 000個細胞表面抗原的表達情況。以CD45/SSC設(shè)門,按五色免疫熒光直接標記法測定骨髓有核細胞各種抗原的表達,分別檢測細胞的CD10、CD19、CD5、CD13、CD33、HLA-DR、CD38、CD34、CD16、CD11b、CD36、CD64、CD56、CD14、CD20、CD2、CD3、CD4、CD7、CD8、CD22、CD24、cTDT、cCD79a、cCD22、cCD3、cMPO、IgM、CD79b、CD45、CD61等抗原的表達。所用抗體以及溶血劑、破膜劑購自貝克曼-庫爾特公司,儀器為貝克曼FC-500MPL。
1.2.4 RT-PCR檢測融合基因 Trizol法提取細胞總RNA,采用多重巢式RT-PCR技術(shù),定性檢測43種常見的白血病融合基因。使用日本Thermo 公司PTC-200 型PCR 儀,試劑盒為科研用白血病融合基因檢測試劑盒(上海源奇生物公司)。檢測的基因包括MLL/AF4、TEL/AML1、SIL/TAL1、ETV6/RUNX1、dupMLL、MLL/ENL、E2A/PBX1、SET/CAN、BCR/ABL、TLS/ERG、E2A/HLF、CALM/AF10、HOX11L2、HOX11、MLL/AF10、NPM/ALK等。
1.2.5 染色體核型分析 取肝素鈉抗凝新鮮骨髓液約3 mL,應(yīng)用植物血凝素進行短期培養(yǎng)(24 h)后立即制片,收集骨髓細胞中有絲分裂中期細胞行吉姆薩染液染色,經(jīng)G顯帶于染色體分析儀上,行骨髓染色體核型分析。
2 結(jié)果
2.1 臨床表現(xiàn) 2例患兒既往行骨髓細胞學檢查排除血液造血系統(tǒng)疾病,并經(jīng)ANA系列等自身抗體譜檢查排除自身免疫性疾病,診斷為ITP。患兒都曾經(jīng)對激素、丙球顯效或部分顯效。患兒1發(fā)病時年齡較小,在新生兒科曾經(jīng)使用丙球、激素治療,出院后予口服潑尼松維持治療,曾經(jīng)一度血小板恢復正常,并予激素減停。初診14個月后,該患兒再次因“發(fā)熱咳嗽5 d,皮膚瘀點瘀斑4 d”入住我科。患兒2在初次診斷時給予靜脈使用地塞米松治療,出院后予口服潑尼松片維持,但其出院后血小板計數(shù)反復減少,約5個月后,因“反復便血22 h”入住我院。
2.2 白血病MICM分型 2例患兒入院后,完善骨髓MICM分型檢查。患兒1的骨髓細胞學即提示AMKL,見圖1。白血病免疫表型:可見約1.10%免疫表型異常髓系原始細胞,表達CD41、CD42b、CD61,原始巨核細胞可能性大,請結(jié)合骨髓形態(tài)學、小巨核酶標等結(jié)果綜合考慮;骨髓染色體:54,XX,+2,+i(6)(p10),+7,+8,+10,+10,+19,+19[16]/46,XX;白血病融合基因篩查及髓系預后基因篩查(包括RUNX1基因)均為陰性。
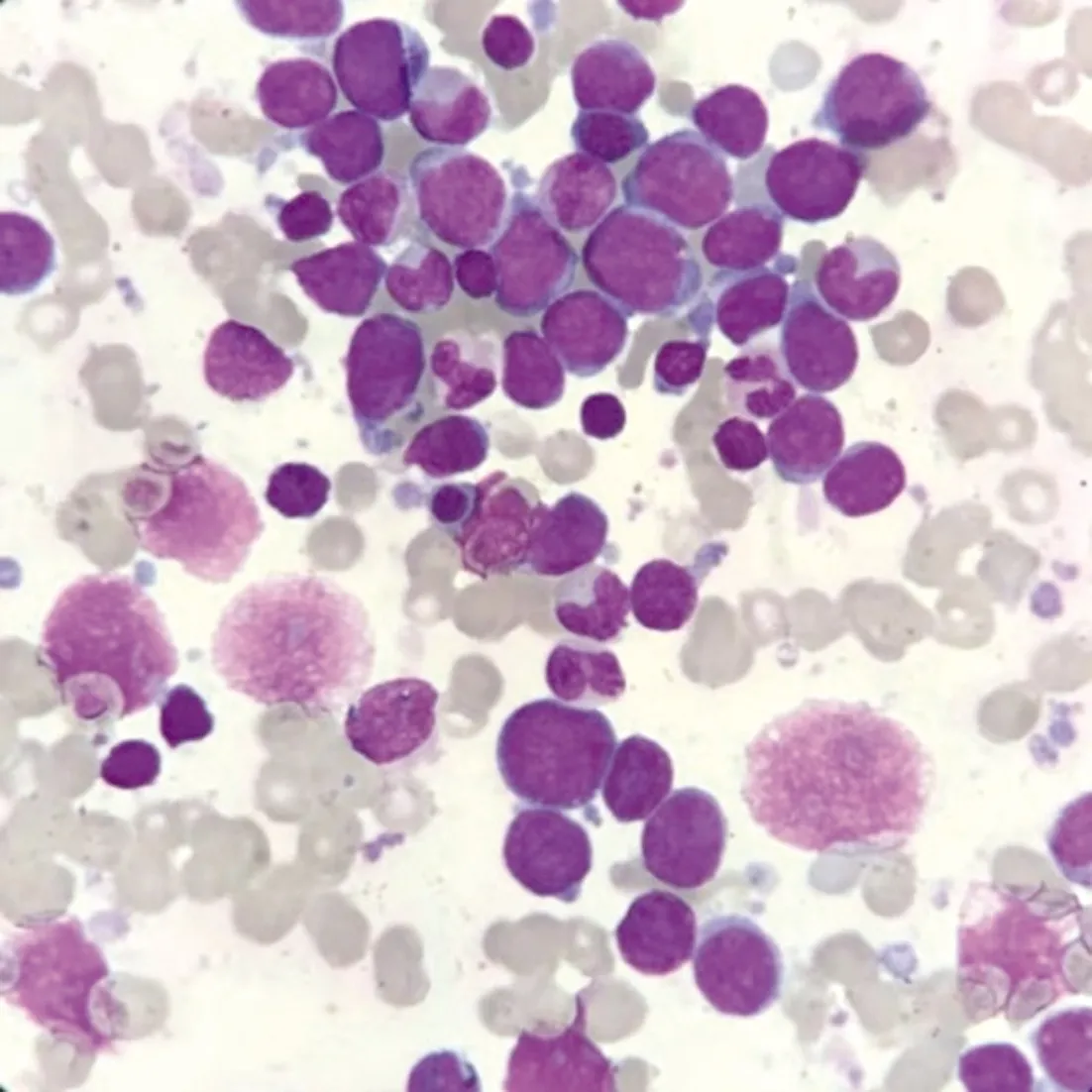
圖1 患兒1骨髓細胞學(瑞-吉姆薩染色×1 000)
患兒2再次診斷時骨髓細胞學形態(tài)并不典型,見圖2。白血病免疫分型:原始細胞占有核細胞總數(shù)約69.4%,該群細胞表達為CD4、CD36、CD41,部分表達為CD7、CD33、CD38、CD61、CD117,少量表達為CD13,不表達CD2、CD3、CD5、CD8、CD10、CD11b、CD14、CD15、CD16、CD19、CD20、CD42b、CD56、CD64,傾向于AML-M7可能性大;骨髓染色體核型為復雜核型:46,XX,der(8)t(8;12)(q24;q21),der(9)t(9;10)(p22;p11.2),-10,der(12)t(10;12)(q11.2;q15),add(17)(p13),-19[7]/46,XX[13];白血病融合基因篩查(包括RUNX1基因)均為陰性。患兒2因經(jīng)濟原因,未行髓系白血病預后基因檢測。
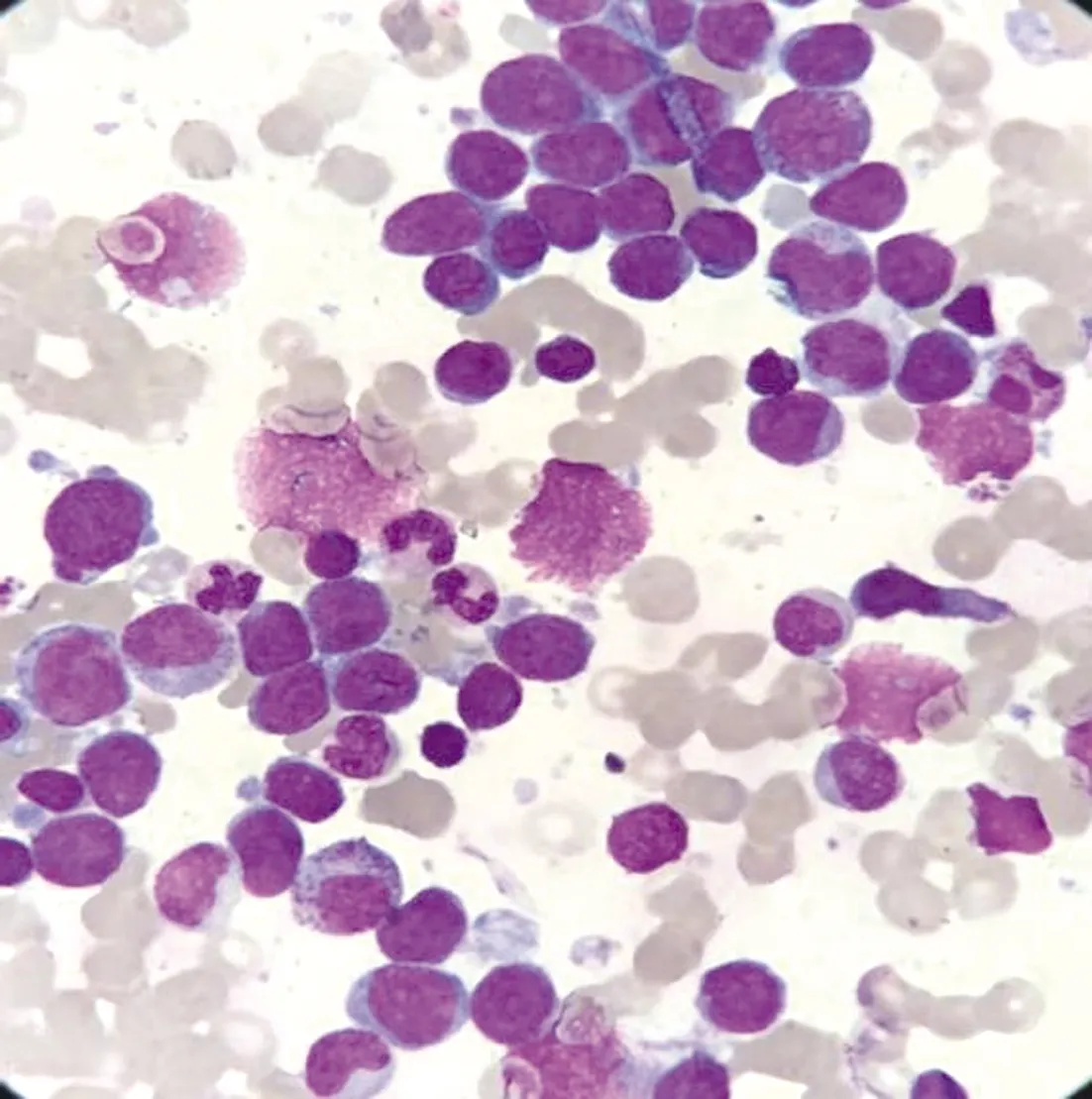
圖2 患兒2骨髓細胞學(瑞-吉姆薩染色×1 000)
2.3 骨髓病理檢查 患兒1在白血病MICM分型檢查后,進行骨髓病理學檢查。骨髓病理結(jié)果:骨髓有核細胞增生程度大致正常(60%);粒/紅比例略高;粒系以偏幼稚細胞散在易見;紅系以中晚幼紅細胞為主;未見明顯巨核細胞;淋巴細胞散在少數(shù);骨髓間質(zhì)未見纖維化。骨髓免疫組化提示:CD34小血管(+);CD117少(+);CD20散在少(+);MPO散在(+);Lyso部分區(qū)域多(+);CD99少(+);CD61巨核細胞(+),為小淋巴樣巨核細胞;E-cad偶見(+)。其骨髓病理及組化染色結(jié)果均支持AMKL診斷。見圖3。
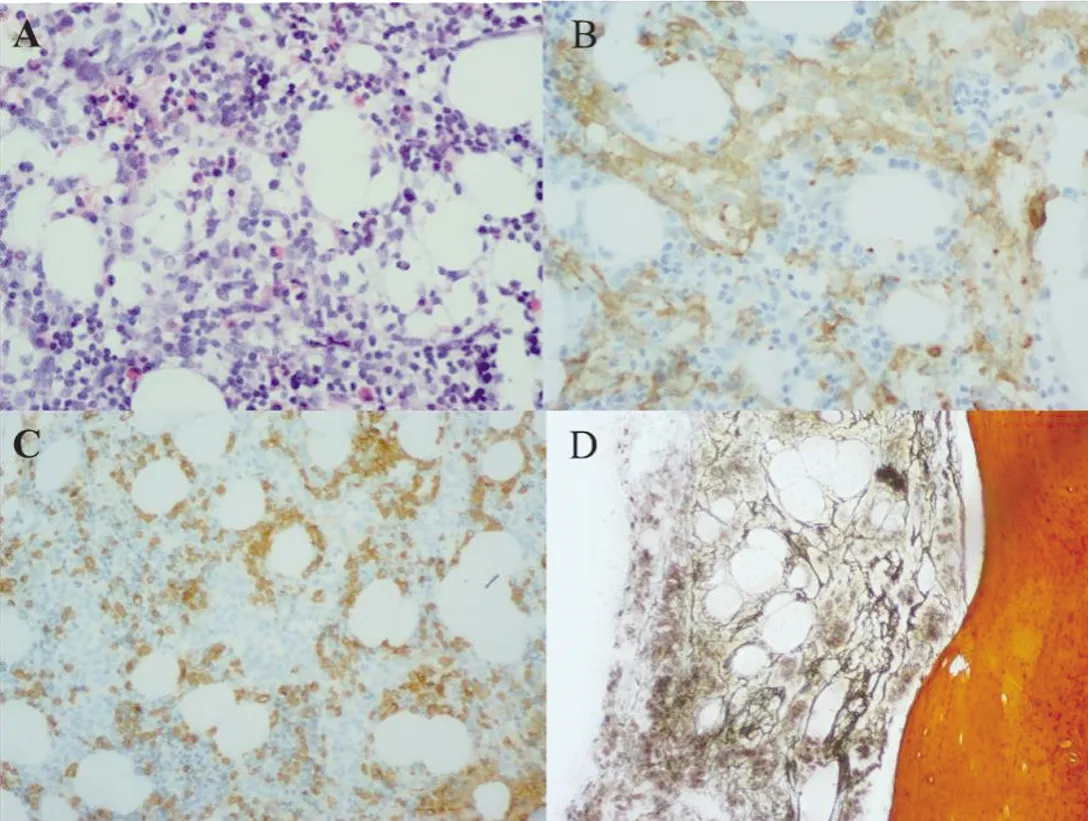
圖3 患兒1骨髓病理檢查
2.4 治療與預后 患兒1入院后,經(jīng)骨髓MICM分型及骨髓病理學檢查,明確診斷為AMKL,進入CCLG-AML2016協(xié)作組方案化療。先后給予誘導化療(DAE方案)、誘導后鞏固化療(IAE方案)、鞏固化療(HA方案)及CLASP方案化療,DAE誘導第28天即達骨髓細胞學完全緩解,已行異基因造血干細胞移植(臍帶血),移植術(shù)后恢復可,目前仍在密切隨訪中。患兒2入院后,經(jīng)骨髓MICM分型診斷為AMKL,最終因經(jīng)濟原因放棄進一步治療,電話回訪時,患兒2已經(jīng)死亡,自其血小板減少起病至死亡時間僅5個月余。
3 討論
ITP 是一類自身免疫性疾病,早在1951 年,Harrington等[4]就已經(jīng)通過實驗驗證了ITP 的病理生理機制為自身抗體引起的自身免疫功能紊亂,導致網(wǎng)狀內(nèi)皮系統(tǒng)過度清除自身血小板。ITP 的發(fā)病機制甚為復雜,其病理生理過程主要由體液免疫及細胞免疫異常介導的血小板生成不足和破壞增多所致[1],涉及細胞因子、細胞程序性死亡、氧化應(yīng)激、感染、妊娠和藥物等諸多方面[5]。無論是成人[6]亦或是兒童[7],目前在ITP的診斷方面主要強調(diào)“排他性”診斷,即排除其他任何已知明確可致血小板減少的疾病,包括遺傳性血小板疾病。本例報告中,初診時2例患兒經(jīng)骨髓細胞學除外血液造血系統(tǒng)惡性疾病及骨髓衰竭性疾病,并經(jīng)自身抗體譜篩查初步排除其他自身免疫性疾病,結(jié)合2例患兒曾經(jīng)對激素、丙球有效,故診斷為ITP。
目前,國內(nèi)外報道血小板減少癥轉(zhuǎn)化為血液造血系統(tǒng)惡性疾病的病例不多,大多轉(zhuǎn)化為髓系惡性疾病,包括AML和骨髓增生異常綜合征(myelodysplastic syndrome, MDS),但大部分見于成人。兒科患者以具有AML傾向的家族性血小板疾病(familial platelet disorder acute myelogenous leukemia, FPD/AML)最為常見[3,8]。FDP/AML絕大部分都以血小板減少和/或出血傾向為首發(fā)癥狀起病,是由位于21q22.1上的RUNX1基因發(fā)生突變所致,可有陽性血液病家族史[9]。據(jù)統(tǒng)計,20%~65%的FDP/AML患者逐漸發(fā)展成為AML/MDS,其轉(zhuǎn)變過程長短不等,已有M1、M2、M4和M5的報道。
研究[9-10]顯示,F(xiàn)DP/AML發(fā)生MDS或AML的高峰年齡約40歲,預后較差,但目前報道的FDP/AML轉(zhuǎn)化為AML的兒科病例亦不在少數(shù)[11-12]。本研究2例患兒均以血小板減少起病,逐漸出現(xiàn)血象其他兩系改變,后經(jīng)骨髓MICM分型確診為AMKL,融合基因檢查證實為RUNX1基因陰性,這在兒童血液專科實屬罕見,目前未見相關(guān)文獻報道。自診斷ITP起病,至AMKL發(fā)病時間,2例患兒分別為14個月和5個月,轉(zhuǎn)化白血病時間較成人更短。遺憾的是,由于經(jīng)濟條件限制,本研究2例患兒皆未能完善遺傳性血小板減少癥相關(guān)基因檢測,這也為進一步研究帶來了困難。
AMKL系一種較為少見的髓系起源急性白血病,在兒童血液專科,非21-三體伴發(fā)的AMKL被認為是一種具有獨特腫瘤生物學特征的白血病亞型,其預后較差,治療強調(diào)早期行造血干細胞移植[13-14]。隨著流式單克隆抗體免疫標記的廣泛應(yīng)用,使得血小板特異性的單克隆或多克隆抗體標記可以識別巨核細胞,1985年FAB協(xié)作組確立了AMKL的診斷標準[15]。本研究患兒1骨髓細胞學、骨髓病理學及流式免疫分型均提示AMKL;而患兒2骨髓細胞學形態(tài)雖不典型,但其白血病免疫分型提示CD41、CD61、CD36等較特異性的巨核系分化抗原均為陽性,故此診斷AMKL亦可成立。AMKL預后兇險,建議在完全緩解后盡早行骨髓移植。本研究患兒1在骨髓完全緩解、配型成功后即行造血干細胞移植,目前仍然存活。
通過本文病例分析,結(jié)合相關(guān)文獻報道,對于反復血小板減少的兒科患者,尤其是小嬰兒,即使病程不長,也應(yīng)密切隨訪骨髓細胞學,及時完善HT相關(guān)基因檢查,達到及時診斷、早期治療的目的。同時,在今后的兒童ITP診療指南中,應(yīng)加入遺傳學檢查的建議,讓更多的患兒得到規(guī)范、合理的診治。
