固相萃取-高效液相色譜法測定保健食品中8種皂苷化合物含量
2020-07-23 16:37:35馬海建王利娟江晨舟邵明華張馳中
江蘇農業學報 2020年3期
關鍵詞:固相萃取
馬海建 王利娟 江晨舟 邵明華 張馳中
摘要:旨在建立一種固相萃取結合高效液相色譜檢測保健食品中8種皂苷化合物(三七皂苷R1與人參皂苷Rg1、Re、Rf、Rb1、Rc、Rb2、Rd)的方法,考察不同溶劑和不同超聲時間對皂苷化合物提取效率的影響以及不同固相萃取小柱對回收率的影響。通過試驗,確定先用30%甲醇溶液超聲20 min進行提取,然后用C18小柱凈化的前處理方法,并對優化后的方法進行方法學驗證。結果表明,優化后的前處理方法+高效液相色譜法操作簡便、耗時少,8種皂苷化合物峰面積與含量線性方程的決定系數均可達到0.999 0以上,檢出限為3.7~11.4 μg/g,方法的精密度和重復性良好,樣品在24 h內穩定,樣品的加標回收率為85.38%~108.41%。該方法可操作性強、穩定性好,可以同時檢測含有三七、人參、西洋參和高麗參等原料的保健食品中8種皂苷化合物含量。
關鍵詞:保健食品;皂苷化合物;固相萃取;高效液相色譜法(HPLC)
中圖分類號:TS207.3文獻標識碼:A文章編號:1000-4440(2020)03-0743-08
Determination of eight saponins contents in health-care food by solid phase extraction-high performance liquid chromatography
MA Hai-jian,WANG Li-juan,JIANG Chen-zhou,SHAO Ming-hua,ZHANG Chi-zhong
(ANPEL Laboratory Technologies
Abstract:The research aims to detect eight saponins including notoginsenoside R1, ginsenoside Rg1, Re, Rf, Rb1, Rc, Rb2 and Rd in health-care food by the method of solid phase extraction-high performance liquid chromatography (SPE-HPLC). Effects of different solvents and ultrasonic time on the extraction efficiency of saponins and the influence of different solid phase extraction columns on the recovery rate were investigated. The pretreatment method was determined that ultrasonic extraction was performed by using 30% methanol solution for 20 minutes, and then the C18 solid phase extraction column was used for purification. The methodology validation was applied to evaluate the optimized methods. The results showed that the optimized pretreatment method combined with HPLC was easy to operate and took less time. The determination coefficients of peak area and content of eight saponins were all above 0.999 0 and the detection limit was 3.7-11.4 μg/g. The method showed good precision and reproducibility, and the sample was stable within 24 hours. The recovery rate of standard addition was 85.38%-108.41%. The method was stable and easy to operate, and could be used to detect the contents of eight saponins in health-care food using Panax notoginseng, Panax ginseng, Panax quinquefolius as raw materials.
Key words:health-care food;saponins;solid phase extraction;high performance liquid chromatography(HPLC)
隨著中國經濟的快速發展與人們生活水平的不斷提高,人們的健康意識和向往健康生活的心態也在逐漸提升,保健食品市場得到快速發展。目前,中國保健食品的產值約有4.000×1011元[1],其中被宣傳能夠增強人的免疫力和緩解疲勞類保健食品一直是中國市場上占比較高的一類產品,這類保健食品多以名貴中藥材為原料,如人參、西洋參和三七等[2-3],這些原料的成本相對較高,導致保健食品市場上存在一些以假亂真、以次充好的現象。
據《中華人民共和國藥典一部》(2015年版)[4]記載,人參、西洋參的主要活性標志成分有人參皂苷Rg1、Re、Rf、Rb1、Rc、Rb2和Rd,三七的主要活性標志成分有三七皂苷R1與人參皂苷Rg1、Re、Rb1、Rd。這些皂苷類化合物具有耐缺氧、抗衰老和提高機體免疫力等作用[5-7]。目前,針對皂苷的檢測方法有比色法、薄層色譜法、高效液相色譜法(High performance liquid chromatography,HPLC)、液相色譜質譜聯用法(Liquid chromatograph mass spectrometer, LC-MS)等[8-9]。其中,比色法存在操作繁瑣、專屬性差、耗時長、測定結果干擾因素多等缺陷[10-11]。相關標準和文獻中的高效液相色譜法則主要針對單一來源產品中幾種皂苷化合物的檢測[12-13],主要采用加熱回流法或超聲法提取、大孔吸附樹脂柱凈化,但是凈化柱手工裝填過程繁雜且重復性較差,而且保健食品可能會使用多種原料,進一步增加了操作的復雜性。為了更好地評價保健食品中皂苷成分的含量,本研究擬建立用于檢測三七皂苷R1與人參皂苷Rg1、Re、Rf、Rb1、Rc、Rb2、Rd共8種皂苷化合物的HPLC方法,同時對前處理條件進行優化,以期為含有三七、人參類保健食品中皂苷化合物的科學檢測提供參考。
1材料與方法
1.1試驗材料
含有人參、西洋參、高麗參、三七的保健食品,購于上海市某連鎖藥店、上海市某大型購物超市及電商平臺。共購買7種保健食品作為試驗樣品,其中包含1種片劑類樣品(某品牌的西洋參含片)、3種膠囊類樣品(某品牌的西洋參膠囊、某品牌的三七西洋參膠囊和某品牌的三七丹參膠囊)、3種口服液類樣品(某品牌的人參蜂王漿口服液、某品牌的高麗紅參飲品和某品牌的高麗參口服液),以上樣品均在其產品保質期內且外觀完整無破損。
1.2試劑與儀器
標準品:純度>99%的三七皂苷R1與人參皂苷Rg1、Re、Rf、Rb1、Rc、Rb2、Rd,均由上海安譜實驗科技股份有限公司提供。
試劑與耗材:甲醇(色譜純,CNW)、乙醇(色譜純,CNW)、正丁醇(色譜純,CNW)、乙腈(色譜純,CNW)、磷酸(色譜純,CNW)、CNWBOND C18 SPE小柱(500 mg,6 ml)、CNWBOND 親水疏水平衡(Hydrophilic-lipophilic banlanced copolymer, HLB) SPE小柱(500 mg,6 ml)、CNWBOND 大孔吸附樹脂XAD-2 SPE小柱(4 g,10 ml)均由上海安譜實驗科技股份有限公司提供。試驗用水為筆者所在實驗室自制的純凈水。
儀器與設備:Thermo U3000高效液相色譜儀(美國賽默飛世爾科技公司產品),2300TH型數控超聲波清洗器(頻率為40 kHz, 功率為100 W,上海安譜實驗科技股份有限公司產品),24位水浴氮吹儀(上海安譜實驗科技股份有限公司產品),TDL-40C臺式大容量離心機(上海安亭科學儀器廠產品)。
1.3試驗方法
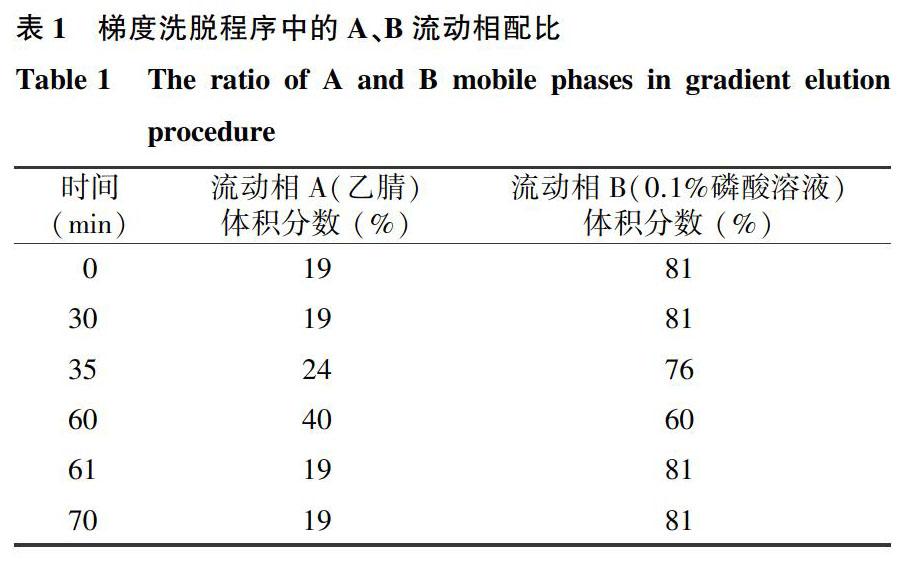
1.3.1色譜條件參照《中華人民共和國藥典一部》(2015年版)中人參總皂苷部分的檢測方法[4],并略作修改。色譜柱為CNW Athena C18 (4.6 mm×250.0 mm, 5 μm),柱流速為1.3 ml/min,柱溫為30 ℃,進樣量為10 μl,紫外檢測器波長為203 nm,流動相A為乙腈,流動相B為0.1%磷酸溶液,梯度洗脫。具體流動相配比見表1。
1.3.2標準品溶液的制備分別精確稱取8.656 19 mg三七皂苷R1與9.336 71 mg人參皂苷Rg1、10.031 90 mg人參皂苷Re、9.024 84 mg人參皂苷Rf、11.498 50 mg人參皂苷Rb1、10.162 10 mg人參皂苷Rc、9.458 62 mg人參皂苷Rb2和8.149 60 mg人參皂苷Rd標準品置于10 ml容量瓶中,用甲醇溶解并定容,制得8種皂苷化合物混合標準貯備液,其質量濃度分別為865.62 μg/ml、933.67 μg/ml、1 003.19 μg/ml、902.48 μg/ml、1 149.85 μg/ml、1 016.21 μg/ml、945.86 μg/ml和814.96 μg/ml。分別移取適量體積的標準貯備液,用甲醇稀釋并定容,得到系列標準工作溶液。
1.3.3樣品溶液的制備將片劑類樣品取出后研磨成粉末,混勻備用;將膠囊類樣品的內容物取出,混勻備用;將口服液類樣品移取到樣品瓶中,混勻備用。稱取2 g左右的樣品于離心管中,加入10 ml 30%甲醇水溶液,在室溫下超聲(頻率為40 kHz,功率為80 W)處理20 min,于4 000 r/min離心5 min,轉移上清液至20 ml容量瓶中,用30%甲醇水溶液定容,取5 ml提取液進行下一步的凈化。
將C18固相萃取小柱裝于固相萃取裝置上,加10 ml純甲醇進行活化,再加10 ml純凈水進行平衡,待水流至篩板表面時加入5 ml提取液,控制流速在3 ml/min左右,待提取液流至篩板表面時,加入10 ml純凈水進行淋洗,待淋洗液流完后,放入離心管接收洗脫液,用12 ml純甲醇進行洗脫,控制流速在3 ml/min左右。將洗脫液于50 ℃水浴氮吹至2 ml左右時轉移至5 ml容量瓶中,用甲醇定容,過0.22 μm針式濾器后上機檢測。
1.4數據處理
本試驗數據均用Excel 2010軟件進行分析處理。
2結果與分析
2.1樣品前處理方法的優化
2.1.1提取溶劑的對比皂苷化合物一般以醇(甲醇、乙醇和正丁醇)、純凈水或兩者的混合物為提取液[14]。某品牌的三七西洋參膠囊樣品的預試驗檢測結果表明,該樣品中不含有人參皂苷Rf。為了同時分析對比8種皂苷化合物的提取效率,優化試驗中在樣品粉末表面添加了適量8種皂苷混合標準品并靜置混勻,以保證樣品中均含有8種待測皂苷化合物,然后在不同前處理條件下進行檢測,以對比優化前處理條件。圖1比較了常見的3種溶劑甲醇、乙醇和正丁醇對8種皂苷的提取效果,3種溶劑均為色譜純,含量在99.9%以上。可以看出,用3種不同溶劑提取時,樣品中8種皂苷化合物的提取量無明顯差異,表明使用3種溶劑超聲提取均可較好地完成保健食品中8種皂苷的提取。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd;n=3。
圖2比較了不同濃度[0(純凈水)、30%、70%和100%]甲醇溶液對三七西洋參膠囊樣品中8種皂苷化合物提取效率的影響,可以看出,不同濃度甲醇溶液均可較好地提取樣品中的8種皂苷類目標化合物,當用純凈水提取時,樣品中人參皂苷Rg1和Re的提取量略大于用30%、70%、100%甲醇溶液提取,而人參皂苷Rb1、Rc、Rb2和Rd的提取量則小于用30%、70%、100%甲醇溶液提取;分別用30%、70%和100%甲醇溶液提取的樣品中8種皂苷化合物的量無明顯差異。從8種皂苷化合物的總提取量看出,含甲醇提取溶劑的提取效率要高于純凈水溶劑,考慮到后續試驗要使用C18固相萃取小柱,提取溶劑中甲醇等有機溶劑含量過高會影響小柱的吸附效能,因此最終選擇30%甲醇溶液作為提取溶劑,既保證了提取效率,又可確保在后續固相萃取富集凈化過程中8種皂苷化合物能夠被小柱填料吸附。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd;n=3。
2.1.2超聲條件的對比提取皂苷化合物一般使用加熱回流法,耗時長、提取效率低,而超聲波能有效破碎細胞壁,釋放細胞內容物,具有耗時少、提取效率高、成分不易分解破壞等優點[15]。本研究選用超聲波提取8種皂苷化合物,減少了提取時間,提高了提取效率。為了研究不同超聲時間和超聲次數對8種皂苷化合物提取量的影響,以30%甲醇溶液作為提取溶劑,分別于室溫超聲處理5 min、10 min、20 min和30 min(頻率為40 kHz,功率為80 W),對比不同超聲時間下8種皂苷化合物提取量。同時,對比超聲20 min提取2次、合并2次提取溶液的方法對8種皂苷化合物提取效果的影響。從圖3可以看出,在超聲時間5~20 min,隨著超聲時間的增加,8種皂苷化合物的提取量呈逐漸增大的趨勢。在超聲20 min、30 min和超聲20 min提取2次3種條件下,8種皂苷化合物的提取量無顯著差異。經綜合分析,選擇超聲20 min提取1次作為保健食品中8種皂苷化合物的提取方式。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd;n=3。
2.1.3不同SPE小柱對8種皂苷化合物加標回收率的對比在皂苷化合物的提取物中,常有糖類、鞣質等親水性較強的成分,給皂苷化合物的分離純化增加了難度[16]。本研究選用XAD-2、C18、HLB 3種固相萃取小柱對保健食品的提取液進行凈化,并對凈化效果和加標回收率進行對比。XAD-2小柱填料是一種非離子型網狀苯乙烯-二乙烯基苯基體的吸附樹脂,類似于《保健食品檢驗與評價技術規范》(2003年版)中人參皂苷檢測部分使用的大孔吸附樹脂[16];C18小柱具有較好的疏水性,對皂苷類化合物有較好的富集作用,同時也可除去一些多糖類等親水性強的雜質;HLB小柱基質是由親脂性二乙烯苯和親水性N-乙烯基吡咯烷酮2種單體按比例聚合而成的大孔共聚物,是一種親水-親脂平衡性吸附劑。從圖4可以看出,使用C18小柱和HLB小柱的樣品中8種皂苷的加標回收率(80%~110%)要明顯高于使用XAD-2小柱的加標回收率(30%~60%)。經小柱分步收集和減少上樣量試驗的驗證,推測這可能是因為樣品中的皂苷化合物含量較高,導致XAD-2小柱過載,未能完全吸附目標化合物。傳統皂苷凈化方法使用的大孔吸附樹脂多為手工裝填的層析柱,其填料裝樣量大,保證了目標化合物不會過載,但是手工裝填操作復雜,耗時長,且重復性相對較差。樣品譜圖對比分析可知,C18小柱的除雜效果要略好于HLB小柱。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd;n=6。
圖5為某品牌含三七原料的膠囊樣品用C18小柱凈化前后的色譜結果,可以看出,C18小柱對樣品有相對較好的凈化作用,對色譜柱和儀器有一定的保護作用,并且可以保證較高的回收率。因此,本試驗最終選用C18固相萃取小柱作為保健食品中8種皂苷化合物檢測過程中的凈化柱。
1:未經C18小柱凈化的譜圖;2:經過C18小柱凈化的譜圖。a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rb1;e:人參皂苷Rc;f:人參皂苷Rb2;g:人參皂苷Rd;n=6。
2.1.4不同前處理方法的對比為了驗證本試驗建立的30%甲醇溶液超聲提取C18 SPE小柱凈化方法的提取效率,參照《中華人民共和國藥典一部》(2015年版)人參相關部分中人參皂苷檢測的加熱回流提取方法,與本試驗建立的前處理方法進行提取效率的對比。《中華人民共和國藥典一部》(2015年版)的具體方法如下:稱取1.0 g樣品于索式提取器中,加三氯甲烷加熱回流3 h,棄去三氯甲烷溶液;將殘渣中的溶劑揮發干后移入錐形瓶中,精確加入50 ml水飽和正丁醇,密塞,放置過夜,超聲(頻率為40 kHz,功率為80 W)30 min,過濾,棄去初濾液,精確量取25 ml續濾液,蒸干后用甲醇溶解殘渣并將其轉移至5 ml容量瓶中,加甲醇定容,過0.22 μm針式濾器后上機檢測[4]。
由圖6可以看出,在2種提取方法下,8種皂苷化合物的提取量差別相對較小,在傳統加熱回流提取方法下,人參皂苷Rf、Rc、Rd的提取量略高于本試驗建立的提取法,而三七皂苷R1、人參皂苷Re和Rb2的提取量則略低于本試驗建立的提取法,2種方法提取的人參皂苷Rb1的含量無明顯差異。總體而言,本試驗建立的提取法與傳統加熱回流提取法的提取效率無明顯差異,并且本試驗建立的提取法可以減少有機溶劑的使用量、縮短前處理時間,相比于傳統加熱回流方法,提取效率更高。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd;n=6。
2.2超聲提取-C18小柱凈化方法的方法學考察
2.2.18種皂苷化合物峰面積與質量濃度的線性關系和檢出限取配制好的一系列不同質量濃度的8種皂苷化合物混合標準溶液進行測定。如圖7所示,8種皂苷化合物的峰形良好且均可得到有效分離。8種皂苷化合物峰面積與質量濃度的線性方程、線性范圍、檢出限和定量限見表2,以各組分的峰面積為縱坐標、質量濃度為橫坐標繪制標準曲線發現,8種皂苷化合物均有較好的線性關系,R2均在0.999 0以上。以某品牌高麗紅參飲品為空白樣品,將稀釋后的混合標準品溶液加入空白樣品中,使其加標濃度約為10 μg/g,平行檢測12個樣品,基于響應值標準偏差和標準曲線斜率法測定方法檢出限(Limit of detection, LOD)和方法定量限(Limit of quantitation, LOQ)。結果表明,檢出限為3.7~11.4 μg/g,定量限為12.3~38.0 μg/g,可以滿足保健食品中8種皂苷化合物的檢測要求。
a:三七皂苷R1;b:人參皂苷Rg1;c:人參皂苷Re;d:人參皂苷Rf;e:人參皂苷Rb1;f:人參皂苷Rc;g:人參皂苷Rb2;h:人參皂苷Rd。
Y1~Y8為峰面積,x1~x8為各皂苷含量。
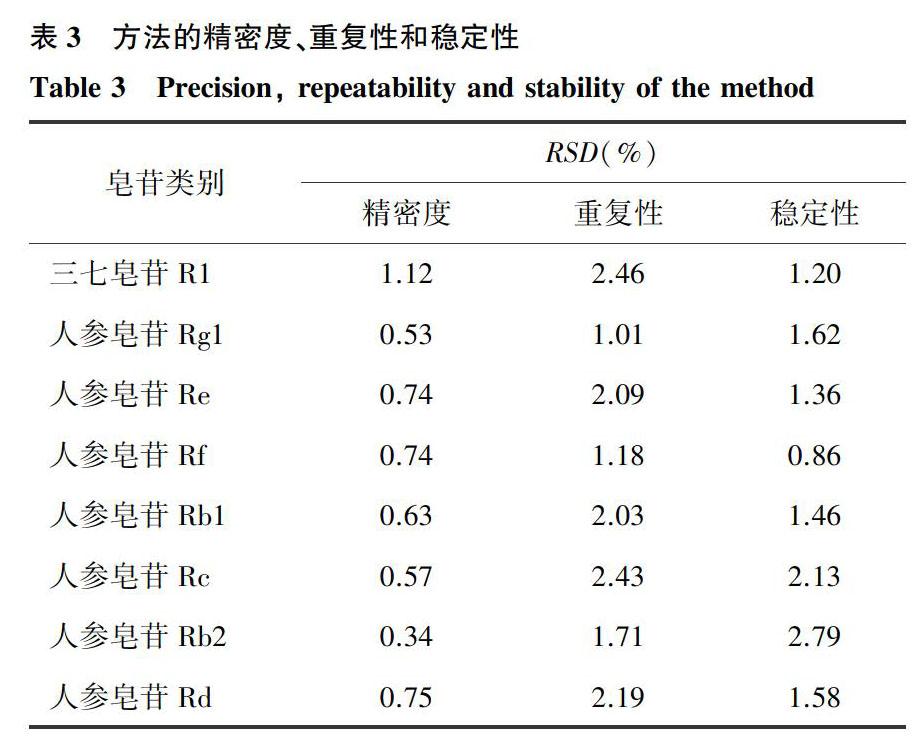
2.2.2方法的精密度、重復性和穩定性取同一份供試品溶液,連續測定6次,由表3可以看出,8種皂苷化合物的相對標準偏差(RSD)為0.34%~1.12%,表明儀器的精密度良好;取同一批次的樣品重復提取6次,進樣測定結果顯示,樣品的RSD為1.01%~2.46%,表明方法的重復性良好;取同一份供試品溶液分別于0 h、2 h、4 h、8 h、12 h和24 h進樣,8種皂苷化合物的RSD為0.86%~2.79%,表明樣品中的8種皂苷化合物至少在24 h內是穩定的。
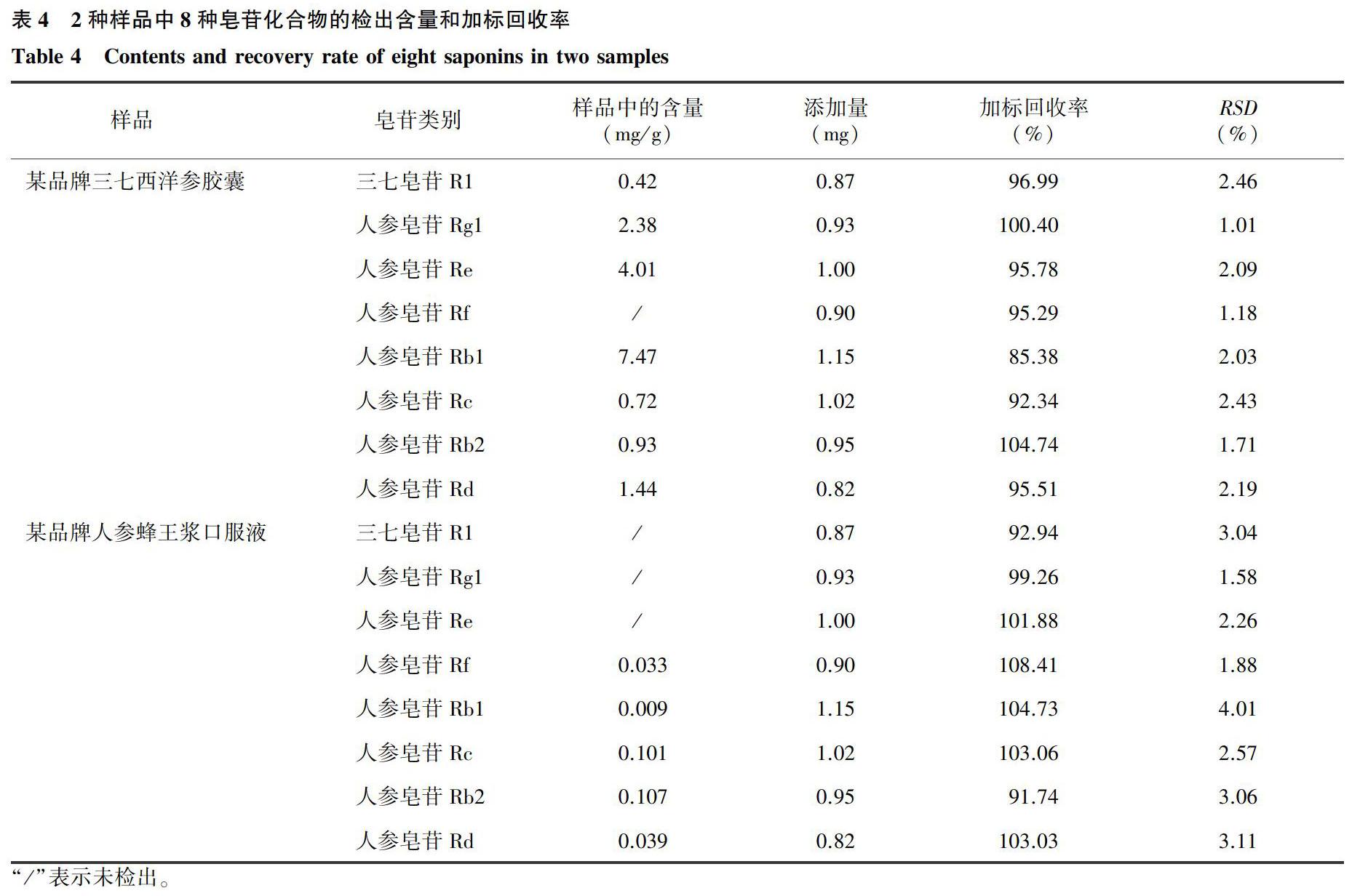
2.2.3加標回收率以某品牌三七西洋參膠囊和某品牌人參蜂王漿口服液作為樣品,添加一定量的混合標準品于待測樣品中,混勻后按照本試驗建立的提取方法提取,并用HPLC法進行檢測,計算8種皂苷化合物的加標回收率。由表4可以看出,三七西洋參膠囊樣品中8種皂苷化合物的加標回收率為85.38%~104.74%,人參蜂王漿樣品中8種皂苷化合物的加標回收率為91.74%~108.41%,2種樣品的RSD均在5%以下,滿足檢測要求。綜合分析可知,本試驗建立的提取方法+HPLC法可應用于保健食品中三七皂苷R1與人參皂苷Rg1、Re、Rf、Rb1、Rc、Rb2、Rd這8種皂苷化合物含量的檢測。
2.3市售多種樣品的檢測
按照上述研究確定的方法,對市場上7種保健食品中的皂苷化合物進行檢測,這些樣品的原料包括人參、西洋參、高麗參和三七等。由表5可以看出,從樣品4和樣品5這2種含有三七的產品中均檢出三七皂苷R1;從樣品1、樣品2、樣品4、樣品5和樣品7中檢測到的8種皂苷化合物的總含量與產品包裝上標注的總皂苷含量基本一致;用本方法從樣品3和樣品6中檢測到的8種皂苷總含量要小于其標簽中標注的總皂苷含量。
3結論
本研究通過比較不同提取溶劑、超聲時間、提取次數、固相萃取小柱對保健食品中8種皂苷化合物提取量的影響,優化了以人參、西洋參、高麗參和三七等為主要原料的保健食品中8種皂苷化合物的提取方法。方法學驗證結果表明,該方法的線性關系較好,精密度和重復性良好,樣品至少在24 h內穩定,樣品的加標回收率為85.38%~108.41%。此外,該方法操作簡便、耗時少、重現性好,可以滿足以人參、西洋參、高麗參和三七等為主要原料的保健食品中8種皂苷化合物的含量檢測。本研究結果為中國保健食品行業更加準確和高效的質量檢測和質量控制提供了依據,有助于保健食品行業更加規范地發展。
“/”表示未檢出。
參考文獻:
[1]張雪艷,王素珍. 保健食品市場亂象成因分析及對策[J]. 中國食品藥品監管, 2018(8): 49-53.
[2]中華人民共和國國家衛生和計劃生育委員會. 食品安全國家標準保健食品: GB16740-2014[S]. 北京: 中國標準出版社, 2014.
[3]鐘文潔,劉淑聰. 保健食品注冊及消費市場現狀分析[J]. 中國藥事, 2016, 30(11): 1056-1062.
[4]國家藥典委員會. 中華人民共和國藥典一部[M]. 北京: 中國醫藥科技出版社, 2015.
[5]YUAN C S, WANG C Z, WICKS S M, et al. Chemical and pharmacological studies of saponins with a focus on American ginseng[J]. Journal of Ginseng Research, 2010, 34(3): 160-167.
[6]WONG A S T, CHE C M, LEUNG K W. Recent advances in ginseng as cancer therapeutics: a functional and mechanistic overview[J]. Natural Product Reports, 2015, 32(2): 256-272.
[7]潘瑩. 人參加工品中有效成分的定量分析及其抗疲勞活性的初步研究[D]. 長春:吉林農業大學, 2013.
[8]張玉婷,馮克然,曹進, 等. UPLC法評價多種人參提取物中人參皂苷的含量[J]. 食品科學, 2013, 34(24): 102-106.
[9]陳朝. 三七及其制劑中皂苷類成分含量測定方法研究概述[J]. 中醫藥導報, 2017, 23(5): 49-53.
[10]李金花,馮有龍,張再平,等. 一測多評法測定西洋參類保健食品中9個皂苷類成分的含量[J]. 藥物分析雜志, 2018, 38(12): 2160-2166.
[11]郭建博,宋莉,呂卓,等. 保健食品中人參皂苷類成分的快速檢測及應用[J]. 食品安全質量檢測學報, 2019, 10(1): 117-122.
[12]徐燦輝,何維為. 西洋參保健食品中7種人參皂苷的高效液相色譜法測定[J]. 食品與藥品, 2015, 17(4): 273-277.
[13]吳曉云,劉春霖,刁飛燕,等. 高效液相色譜法測定三七類保健食品中6種皂苷的含量[J]. 食品安全質量檢測學報, 2017, 8(11): 4412-4417.
[14]郭健,李敏,高巖. UPLC法測定保健品中五種人參皂苷的含量[J]. 中國衛生檢驗雜志, 2012, 22(7): 1507-1509.
[15]李金花,馮有龍,張再平,等. 多指標正交試驗優化美國洋參膠囊中9種人參皂苷類成分測定的前處理方法[J]. 中南藥學, 2017(12): 1757-1760.
[16]彭維,李雙祁,歐愛芬. 不同前處理方法對保健食品中總皂苷含量測定的影響[J]. 食品研究與開發, 2014, 35(8): 90-93.
(責任編輯:徐艷)
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
食品安全導刊(2015年10期)2015-10-26 04:44:22
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
肉類研究(2015年1期)2015-04-08 12:37:48
