基于分組貝葉斯排序的藥物-靶標關系預測
2020-08-03 10:05:52丁棋梁石澤智李建華
計算機工程與應用 2020年15期
丁棋梁,石澤智,李建華
華東理工大學 信息科學與工程學院,上海 200237
1 引言
計算機輔助藥物設計是一個跨學科的研究領域,包括對生物學、化學、物理學和信息學的研究,其目的是加速藥物研發過程。藥物研發的關鍵是尋找藥物和靶標間是否存在相互作用關系(Drug-Target Interaction,DTI)。盡管可以通過體內外測定[1]藥物和靶標間是否存在相互作用,但這些方法時間長且成本昂貴[2]。因此,可以利用計算機技術來預測可能的DTI,通過實驗來篩選藥物[3],可以顯著降低向市場推出新藥的成本[4]。
目前主要有兩類計算機預測DTI方法:對接模擬和機器學習方法。對接模擬方法[5]是利用靶標的3D結構來鑒定與藥物是否存在潛在結合位點,但是非常耗時且需要靶標的3D結構,并不是所有的靶標都具有3D結構。一些研究人員報告,標準的分子對接評分函數可能被基于機器學習的評分函數所取代,并具有改進的預測結果[6]。機器學習方法通常利用藥物和靶標結構的特征[7]、藥物的副作用[8]以及已經確認的DTI的知識[9]。
近年來,機器學習技術的迅速發展為預測DTI提供了有效的方法,基于機器學習的方法大致分為四類:分類方法、矩陣分解方法、核方法和網絡推理方法。支持向量機(SVM)是一種經典的分類方法,目前已經被Nagamine[10]和Wang等人[11]使用來預測DTI。矩陣分解的兩種代表性方法是雙核的核化貝葉斯矩陣分解(KBMF2K)[12]和多相似協同矩陣分解(MSCMF)[13]。核方法主要包括藥物-靶標對核方法(PKM)[14]、網絡拉普拉斯正則化最小二乘法(NetLapRLS)[15]和具有Kromecker積核的正則化最小二乘法(RLS-Kron)[16]。Bleakley和Yamanishi[17]建立了二部局部模型(BLM),并對藥物-靶標相互作用網絡進行學習,是一種典型的網絡推理方法。然而,這些基本方法都沒有預測新藥物或新靶標的能力。為了預測新藥物或新靶標,Mei等人[18]和Laarhoven等人[19]通過交互鄰居信息解決了這個問題。
前面提到的方法側重于預測所有未知藥物-靶標對是否存在相互作用的概率,導致時間復雜度較高。為了降低時間復雜度,Ladislav等人[20]提出了一種新的思路,以藥物為中心進行研究,分別對特定藥物存在相互作用的靶標進行排序。靶標排名越靠前就最有可能與該藥物存在相互作用,并根據預測的相互作用概率分別為每個藥物確定未知靶標。他們使用貝葉斯個性化排序的矩陣分解技術(BPR-MF)來預測DTI,稱為貝葉斯排序方法(Bayesian Ranking,BR)。雖然BR取得了很好的效果,但該方法的局限是所有藥物間是相互獨立的,無法使一些相似的藥物產生互動。根據與特定靶標存在相互作用的藥物間是存在相似性的現實,為了使這些相似的藥物間產生互動,本文對這些相似藥物進行了分組,并推導出分組貝葉斯排序的理論模型。最后通過實驗驗證,提高了其性能。
2 原理及相關工作
在這一部分,首先描述了在研究中使用的數據集,以及獲得藥物和靶標相似度矩陣的過程。然后詳細描述了藥物-靶標預測問題,并在DTI預測的背景下引入貝葉斯排序方法。最后詳細描述采用貝葉斯排序方法對藥物-靶標關系預測的優勢。
2.1 原理
本文使用了五個公開的藥物-靶標相互作用數據集,即核受體(NR)、G蛋白偶聯受體(GPCR)、離子通道(IC)、酶(E)和激酶(Kinase)。表1給出了每個數據集的一些統計數據,包括藥物總數、靶標總數、已知相互作用總數和最近驗證的相互作用總數。

表1 數據集統計
每個數據集包含三個矩陣:(1)藥物-靶標相互作用矩陣;(2)藥物相似度矩陣;(3)靶標相似度矩陣。一般來說,藥物相似度和靶標相似度可以用多種方法計算。本文采用與對比方法同樣的算法來計算靶標相似度和藥物相似度,通過序列對齊方法計算靶標相似度,如Smith-Waterman算法。除Kinase數據集外,其余四個數據集都通過SIMCOMP[21]方法計算藥物相似度;而在Kinase數據集中,則通過2D Tanimoto系數計算藥物相似度。
2.2 基本符號及問題描述
在本文中,假設有m個藥物和n個靶標,D是所有藥物的集合,T是所有靶標的集合。藥物與靶標之間的相互作用關系用二元矩陣Y∈Rm×n表示,其中每個元素yij∈{0,1}。如果藥物已經被實驗驗證與靶標存在相互作用,則設為1;否則,設為0。定義新藥物集和新靶標集合TN=藥物相似度矩陣用SD∈Rm×m表示,靶標相似度矩陣用n×n表示。
矩陣分解法的目的是將藥物和靶標映射到一個共享的潛在空間,其中f表示其維數(潛在因子的個數),ui∈Rf表示藥物di的潛在因子,表示靶標tj的潛在因子。定義U∈Rm×f為所有藥物潛在因子的矩陣,V∈Rn×f為所有靶標潛在因子的矩陣。藥物di與靶標tj相互作用的預測概率r?ij定義為其潛在因子的點積,因此最終預測藥物與靶標之間的相互作用關系矩陣Y?可以用Y?=UVT來表示。進一步將每種藥物的訓練集定義為三元組訓練集Ds?D×
本文以藥物為中心的重定位方法預測DTI。主要目標是對任意藥物d∈D,提供所有靶標的排序,排名最靠前的靶標以最大的概率與藥物d產生相互作用。
2.3 貝葉斯排序方法
貝葉斯排序方法是建立在BPR-MF算法三大假設上,而BPR-MF算法參考了文獻[20]。如下是BPR-MF算法基于的三大假設:
(1)藥物和靶標之間的相互作用行為是彼此獨立的。
(2)藥物和靶標的特征矩陣均服從高斯分布,且平均值為0,方差為常數。
(3)藥物-靶標相互作用關系矩陣的預測值和真實值之間的誤差需要滿足均值為0,方差為常數的高斯分布。
本文采用的是貝葉斯排序和矩陣分解的組合方法,并記為BPR-MF,它是建立在3個基礎假設上的。首先基于這些假設建立相應的概率模型,然后利用貝葉斯公式,最大化后驗概率,建立起對應的優化準則,最后對其進行求解獲得相應的藥物和靶標特征矩陣,進而重構藥物-靶標關系網絡進行未知藥物-靶標關系的預測。
為了使每個藥物盡可能找到其所有正確的靶標排序,需要用貝葉斯公式最大化如下后驗概率:
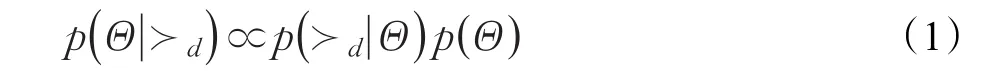
其中,Θ表示矩陣分解參數。基于假設(1)可以得到特定藥物的概率函數p(?d|Θ)用以下公式來表示:

藥物d與靶標tj相互作用的概率大于該藥物與靶標tk相互作用的概率定義如下:
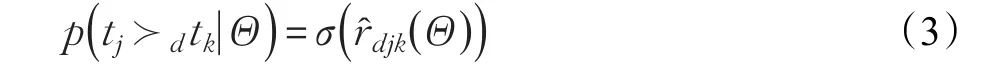

2.4 貝葉斯排序方法的優勢
貝葉斯排序方法的一個核心步驟是構建新的訓練集,不同的是,這里的訓練樣本不是藥物-靶標對,而是一個由藥物和靶標組成的三元組,這里記為(d,ti,tj),其中藥物d與靶標ti存在相互作用關系,而與靶標tj的相互作用關系未知。
貝葉斯排序方法使用三元組作為新的訓練集,與傳統方法進行對比,不再需要對所有未知藥物-靶標對是否存在相互作用關系進行預測,只需要對特定藥物存在相互作用的靶標進行排序。靶標排名越靠前就最有可能與該藥物存在相互作用,并根據預測的相互作用概率分別為每個藥物確定未知靶標,可以明顯降低時間復雜度。
3 分組貝葉斯排序方法
在這一部分,首先描述兩個新定義,然后提出新假設及成立的依據。最后在新假設的基礎上推導出分組貝葉斯排序(Group Bayesian Ranking,GBR)的理論模型,來平滑新藥物和新靶標。
3.1 分組思想介紹
定義1(個體相互作用)個體相互作用是藥物di和靶標tj之間相互作用的概率。例如,藥物di和靶標tj之間相互作用的概率表示為
定義2(分組相互作用)分組相互作用是對特定靶標存在相互作用的藥物集合和該靶標存在相互作用的概率。例如,藥物集合G和靶標tj之間相互作用的概率表示為。其中表示已知與靶標tj存在相互作用的全體藥物集合。
本文在分析本地和遠程生物信息系統模型基礎上,提出了一個基于數據倉庫的架構思想的、適用于病毒序列數據庫的集成系統架構。其目的是實現對病毒序列數據的分類提煉、整理和系統化,并提供相應的集成分析服務。同時以流感病毒序列為例,建立了一個流感病毒序列集成數據庫系統,為相關數據庫的構建積累了一定的經驗。下一步將對更多病毒類別(如腸道病毒、腺病毒等)的數據進行集成,進一步擴充和完善現有的病毒序列集成數據庫系統。
新假設:如果藥物-靶標對(di,tj)是已知存在相互作用關系的,而藥物-靶標對(di,tk)是否存在相互作用是未知的,本文提出的新假設用以下式子進行表示:

其中,且。通過圖1可以更加直觀地推出新假設。藥物d1,d2,d3與靶標t1是已知存在相互作用的,而藥物d1與靶標t2是否存在相互作用是未知的。根據定義 1,都大于r?,所以12r?12也成立,即,得到新假設:(G,t1)?(d1,t2),這里取G={d1,d2,d3}。

圖1 藥物-靶標相互作用關系圖
分組貝葉斯排序方法具體實現步驟如算法1所示。
算法1分組貝葉斯排序方法
輸入:相互作用矩陣Y;相似性矩陣SD,ST;藥物(或靶標)鄰居k的大小。
輸出:更新的相互作用矩陣Y?。
步驟1初始化U,V,b。
步驟2將SD,ST更改為僅包含每個項的前k個最近鄰居。
步驟3使每個藥物靶標對(di,tj),滿足rij=1。
步驟4隨機選擇靶標tk,使rik=0。
步驟5對特定靶標tj存在相互作用的藥物進行隨機分組,使分組大小|G|=1,2,3,4,5。
步驟6 更新bj,bk,ui,vj,vk。
步驟7返回步驟3,直到達到最大迭代次數。
3.2 新假設成立依據
本文根據以下兩個方面信息做出合理的假設:
(1)對于靶標:如果藥物di和靶標tj存在相互作用,且能找到其他藥物對靶標tj也存在相互作用,則藥物di與靶標tj存在相互作用概率大于與靶標tk存在相互作用概率。所以可以用(G,tj)?(di,tk)代替(di,tj)?(di,tk)。
(2)對于藥物:在所有對特定靶標tj存在相互作用的藥物間引入互動是很自然的,因為意味著這些藥物是存在相似關系的。有共同相似關系的藥物組G?Dtrtj,它們對靶標tj都存在相互作用關系。
3.3 理論模型
為了更加精確地研究個體相互作用和分組相互作用對預測結果的不同程度影響,把它們線性地結合起來:
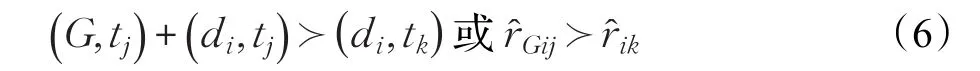
其中,r?Gij=ρr?Gj+(1-ρ)r?ij。 0≤ρ≤1是用于融合兩種不同相互作用的權衡參數,可通過測試驗證集來確定。
有了上述假設,在BR基礎上,用r?Gij代替r?ij,每個藥物就有了新的靶標排序,稱為分組貝葉斯排序。因此,最終分組貝葉斯排序方法目標函數如下:
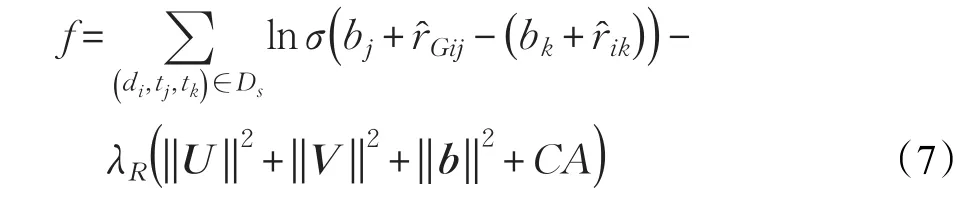
其中,bj和bk分別為靶標tj和靶標tk的偏差,b是所有靶標的偏差,CA是潛在因素距離的正則化項。設訓練集中的一個三元組(di,tj,tk)∈Ds,CA可以用以下公式來表示:

本文使用廣泛的隨機梯度下降(SGD)優化目標函數f,模型參數Θ包括ui,vj,vk,bj和bk。首先需要計算目標函數中參數的梯度,然后根據相應的梯度,模型參數可以更新如下:

3.4 平滑新藥物和新靶標
通過合并鄰居信息進行預測新藥物或新靶標的相互作用。顯然,貝葉斯排序方法不能預測新藥物和新靶標,只能通過負例(未知DTI)了解其潛在因素,這將嚴重破壞整個模型。因此,基于協同過濾的思想[22],本文整合了鄰居信息,得到未知藥物或未知靶標的潛在因素,如下所示:

其中,N+(di)和N+(tj)分別是已知藥物和靶標k個最近鄰居的集合。在實驗中,k通常設置為5以簡化模型。
4 實驗及結果分析
4.1 評價指標
本文采用ROC曲線以下面積(Area Under the ROC Curve,AUC)、標準化折扣累計收益[8](normalized Discounted Cumulative Gain,nDCG)和平均精度(Mean Average Precision,MAP)作為評價指標。AUC值和MAP值幾乎在所有藥物-靶標關系預測中都用來做評價指標,而nDCG值是在最近文獻[8]才提出的評價指標,具有很大的參考價值,所以本文將它納入評價指標。
nDCG通過使用分級相關特征,能區分出具有較高潛在影響的DTI預測。nDCG通過自然截斷僅考慮排名前k個對象對DTI預測所產生的影響,忽略了不重要對象所產生的較小影響,降低了時間復雜度。
4.2 實驗設置及對比方法
為了與先前的研究方法[15,18,20]具有對比性,本文采取了5次10折交叉驗證(CV)的實驗評估了GBR預測方法的性能。并將該方法與5個典型的DTI預測方法進行了比較,如基于高斯核的加權最近鄰[11](WNN-GIP)、協同矩陣分解[13](CMF)、網絡拉普拉斯正則化最小二乘法[15](NetLapRLS)、最近鄰居信息的二部局部模型[18](BLM-NII)和貝葉斯排序方法[20](BR)。
計算每一次交叉驗證的平均值,并重復地運行5次,每次隨機把已知DTI分成10份,得到一個最終的AUC值。并采用同樣的方法計算出nDCG值和MAP值。
4.3 參數設置
在GBR方法中,參數對優化結果起著重要作用。從理論上不難發現,選取的鄰居個數k越多,性能越好,但隨著鄰居個數不斷增加到某個值時,性能改善并不明顯,而時間復雜度將持續提高,導致算法效率低下。在藥物-靶標關系預測中,隨著分組大小||G值增加,性能會越好,但時間復雜度呈現指數級增加。當||G=1時,改進的方法就是BR方法了,所以選取合適的k值和||G值至關重要。
為了深入理解選取鄰居個數和分組大小對GBR方法的影響,將參數調整范圍設置為k∈{3,5,8,15,20,30}和|G|∈{1,2,3,4,5},通過實驗來選取合適的k值和|G|值。從圖2可以看出,當鄰居個數k值大于8時,性能改善并不明顯。從圖3可以看出,當分組大小|G|值大于3時,nDCG改善明顯減低,有些甚至下降。

圖2 鄰居個數的影響
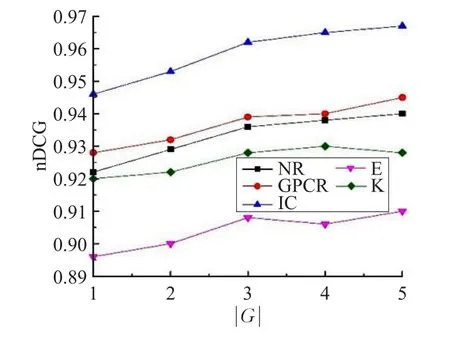
圖3 分組大小的影響
在這個實驗中,將最近鄰的大小設置為k=8,分組大小設置為|G=3|。
4.4 與典型的五種方法進行比較
為了說明本文的GBR方法優于五種典型的DTI預測方法,本文與五種典型方法使用相同公開的數據集,相同的實驗環境。結果匯總在表2~4中,正如所希望的:GBR方法在AUC、nDCG和MAP方面均優于典型方法。

表2 與典型算法的AUC值比較 %

表3 與典型算法的nDCG值比較 %

表4 與典型算法的MAP值比較 %
4.5 預測新的相互作用
這五個公開數據集是幾年前提取的,并一直保持不變,以便于不同方法的比較。然而,這些數據集中一些未知的藥物-靶標對,最近通過生物化學方法已被確認其相互作用,并已上傳到數據庫,如DrugBank[15]、KEGG[23]或Matador[24]。
本文的目的是證明GBR方法比典型方法更能準確地預測最近驗證的藥物-靶標對。若在當前數據庫KEGG、DrugBank或Matador中含有預測的新相互作用,則預測成功。
為了證明GBR方法能夠預測新的相互作用,本文使用了原始數據集的所有相互作用藥物-靶標對,對GBR方法及典型方法進行了訓練,并對非相互作用藥物-靶標對進行了排序。本文只考慮了GPCR和Enzyme數據集中每種藥物的前10個藥物-靶標對。
圖4顯示了在GPCR數據集中的命中數,圖5顯示了在Enzyme數據集中的命中數。正如所期望的,在GPCR數據集中,GBR方法的前10個命中數占總數的63%;在Enzyme數據集中,GBR方法的前10個命中數占總數的34%,明顯高于典型方法。
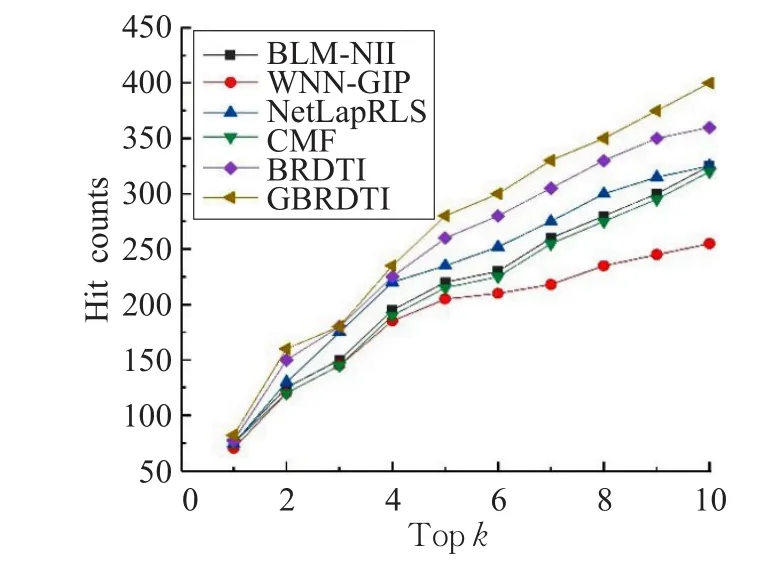
圖4 GPCR數據集上的命中數

圖5 Enzyme數據集上的命中數
最后,表5展示了多種藥物-靶標預測方法在5個數據集上topN的(N=10,30)藥物-靶標關系中預測成功的比例,所有的方法均使用優化后的參數進行實驗。可以看到,GBR方法在所有數據集上的預測準確性都提高了。從橫向看,GBR方法取得了8個最大值,而其他方法最好的情況也才取得3個最大值。
5 結語
本文考慮了分組相互作用對貝葉斯排序方法的影響,根據與特定靶標存在相互作用的藥物具有相似性的現實,把這些相似的藥物進行分組,得到分組的藥物集合。然后根據分組的藥物集合提出新假設,在新假設的基礎上推導出分組貝葉斯排序的理論模型。最后,本文還結合了鄰居信息來平滑新藥物和新靶標的預測。通過相應的實驗,證明了本文方法在性能上優于典型方法。為了將來的工作,計劃開發一種新的對相似靶標進行分組方法以進一步改進性能。

表5 topN個藥物-靶標關系中預測成功的比例
猜你喜歡
兒童時代·幸福寶寶(2022年12期)2022-12-09 11:24:14
中學生數理化·七年級數學人教版(2022年11期)2022-02-14 07:14:12
科普童話·學霸日記(2020年1期)2020-05-08 16:45:11
兒童故事畫報(2019年5期)2019-05-26 14:26:14
小天使·一年級語數英綜合(2019年2期)2019-01-10 11:57:30
兒童繪本(2018年5期)2018-04-12 16:45:32
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
