兒童進(jìn)行性肌營(yíng)養(yǎng)不良臨床特征及基因分析
2020-08-04 11:11:35陳賢娥陳瑯吳麗娟
中國(guó)現(xiàn)代醫(yī)生 2020年15期
關(guān)鍵詞:兒童
陳賢娥 陳瑯 吳麗娟
[摘要] 目的 對(duì)兒童進(jìn)行性肌營(yíng)養(yǎng)不良臨床特征及基因進(jìn)行分析。 方法 納入2013年11月~2019年12月就診本院的進(jìn)行性肌營(yíng)養(yǎng)不良兒童33例,對(duì)其臨床表現(xiàn)、酶譜檢查、肌電圖及基因檢測(cè)等相關(guān)資料進(jìn)行回顧性研究。結(jié)果 33例患兒中有30例(90.9%)為男性,3例(9.1%)為女性,其中11例(33.3%)以行走困難,步態(tài)異常,肢體無(wú)力為主要癥狀,22例(66.7%)以肌酶、肝酶異常升高為首發(fā)癥狀,伴或不伴有智力發(fā)育遲緩。MLPA、NGS-DNA測(cè)序或者肌肉活檢可明確診斷。基因測(cè)序顯示其基因缺失部位主要在2個(gè)區(qū)段,以45~54號(hào)及2~20號(hào)最為常見,但臨床表現(xiàn)與缺失或重復(fù)的片段部位無(wú)太大相關(guān)性。 結(jié)論 認(rèn)識(shí)兒童進(jìn)行性肌營(yíng)養(yǎng)不良的臨床特征及基因突變特點(diǎn),早期進(jìn)行基因篩查,為二胎產(chǎn)前診斷提供借鑒,更好實(shí)現(xiàn)優(yōu)生優(yōu)育,降低本病的發(fā)病率及死亡率。
[關(guān)鍵詞] 進(jìn)行性肌營(yíng)養(yǎng)不良;抗肌萎縮蛋白;兒童;基因篩查
[中圖分類號(hào)] R746.9 ? ? ? ? ?[文獻(xiàn)標(biāo)識(shí)碼] B ? ? ? ? ?[文章編號(hào)] 1673-9701(2020)15-0066-03
Clinical characteristics and genetic analysis of children with progressive muscular dystrophy
CHEN Xian'e ? CHEN Lang ? WU Lijuan ? LIN Xin
Department of Pediatrics, Fujian Provincial Hospital Affiliated to Fujian Medical University, Fuzhou ? 350001, China
[Abstract] Objective To analyze the clinical features and genes of children with progressive muscular dystrophy. Methods A total of 33 children with progressive muscular dystrophy who were treated in our hospital from November 2013 to December 2019 were included, and their clinical manifestations, zymogram, electromyography, and genetic testing were retrospectively studied. Results Of the 33 children, 30(90.9%) were male and 3(9.1%) were female. Among them, 11(33.3%) had difficulty walking, abnormal gait, and limb weakness as the main symptoms, 22(66.7%) had abnormally increased muscle enzymes and liver enzymes as the first symptom, with or without mental retardation. MLPA, NGS-DNA sequencing or muscle biopsy can be used to confirm the diagnosis. Gene sequencing showed that the gene deletion site was mainly in two segments,with 45-54 and 2-20 as the most common.But the clinical manifestations were not much related to the missing or repeated fragments. Conclusion It can provide a reference for the prenatal diagnosis of the second child by recognizing the clinical features and gene mutation characteristics of children with progressive muscular dystrophy and conducting early genetic screening, to realize better outcome of pregnancy and better child-raising, and to reduce the incidence and mortality of this disease.
[Key words] Progressive muscular dystrophy; Anti-dystrophin; Children; Genetic screening
進(jìn)行性肌營(yíng)養(yǎng)不良(progressive muscular dystrophy)是逐漸進(jìn)展的對(duì)稱性肌無(wú)力、肌萎縮、步態(tài)異常為主要表現(xiàn)的原發(fā)性骨骼肌肉病,常見有杜氏進(jìn)行性肌營(yíng)養(yǎng)不良(Duchenne muscular dystrophy,DMD)和貝氏進(jìn)行性肌營(yíng)養(yǎng)不良(Becker muscular dystrophy)等[1]。它是編碼抗肌萎縮蛋白的基因突變引起的X連鎖隱性遺傳病。常見有3種突變類型:缺失突變、重復(fù)突變、微小突變,以缺失突變最常見,絕大多數(shù)是缺失1個(gè)或多個(gè)外顯子[2-3]。這些突變改變基因的閱讀框架,影響抗肌萎縮蛋白的合成,導(dǎo)致肌細(xì)胞的壞死、肌肉組織纖維化,造成肌無(wú)力[4]。通常在3~4歲開始發(fā)病,病情進(jìn)展快,10余歲開始不能行走,一般20~30歲死于呼吸衰竭或心力衰竭[5]。目前尚無(wú)有效治療,因此認(rèn)識(shí)其表型,及早基因篩查,盡早診斷,為產(chǎn)前診斷提供借鑒,以降低本病的發(fā)病率及死亡率。
1 資料與方法
1.1 一般資料
納入2013年11月~2019年12月就診于福建省立醫(yī)院的進(jìn)行性肌營(yíng)養(yǎng)不良兒童33例的病例資料。符合進(jìn)行性肌營(yíng)養(yǎng)不良的臨床表現(xiàn),經(jīng)多重連接依賴式探針擴(kuò)增技術(shù)(Multiplex ligation- dep-endent probe amplification,MLPA)和二代基因測(cè)序(Next generat-ion sequencing technology,NGS)或肌肉活檢明確診斷。
1.2 方法
收集患兒的年齡、性別,臨床表現(xiàn)、酶譜檢測(cè)、肌電圖、心電圖、心臟彩超、基因檢測(cè)如MLPA或NGS、肌肉活檢、家族史、智力情況、治療及隨訪情況。
2 結(jié)果
2.1 病史資料
33例中,男30例(90.4%),女3例(9.1%);確診年齡6個(gè)月~11歲,平均(6.0±3.1)歲。其中11例(33.3%)以行走困難、步態(tài)異常、肢體無(wú)力為主要癥狀,22例(66.7%)以肌酶、肝酶異常升高為首發(fā)癥狀;2例(6.1%)有智力低下,2例(6.1%)有家族史,其中1例是舅舅有類似病史,另1例弟弟也發(fā)病。15例(45.4%)Gowers癥陽(yáng)性;22例(66.7%)雙側(cè)腓腸肌肥大,3例(9.1%)有鴨步態(tài),2例(6.1%)有翼狀肩;33例患者中部分已有不同程度雙下肢肌力下降,1例(3.0%)2級(jí),2例(6.1%)3級(jí),1例(3.0%)4級(jí),2例(6.1%)5級(jí),27例(81.8%)正常。
2.2 輔助檢查
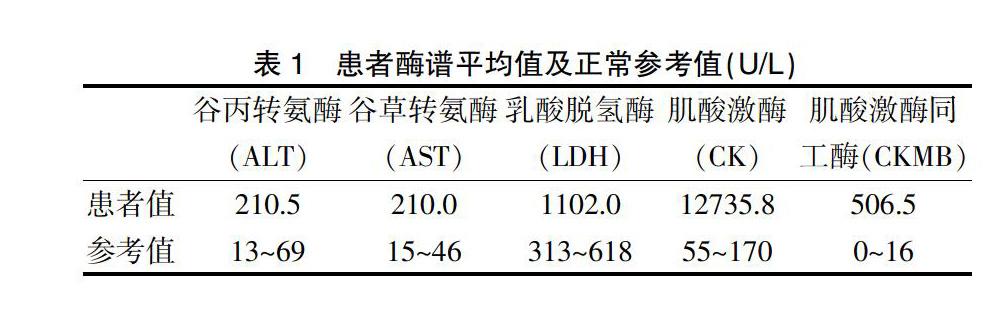
2.2.1 酶譜檢測(cè) ?33例中谷丙轉(zhuǎn)氨酶、谷草轉(zhuǎn)氨酶、乳酸脫氫酶、肌酸激酶、肌酸激酶同工酶均明顯升高,數(shù)值以平均數(shù)(x)表示。見表1。
2.2.2 肌電圖 ?其中25例(75.8%)行肌電圖檢查,有24例示肌源性損害,1例正常;4例拒絕檢查,4例結(jié)果遺失。
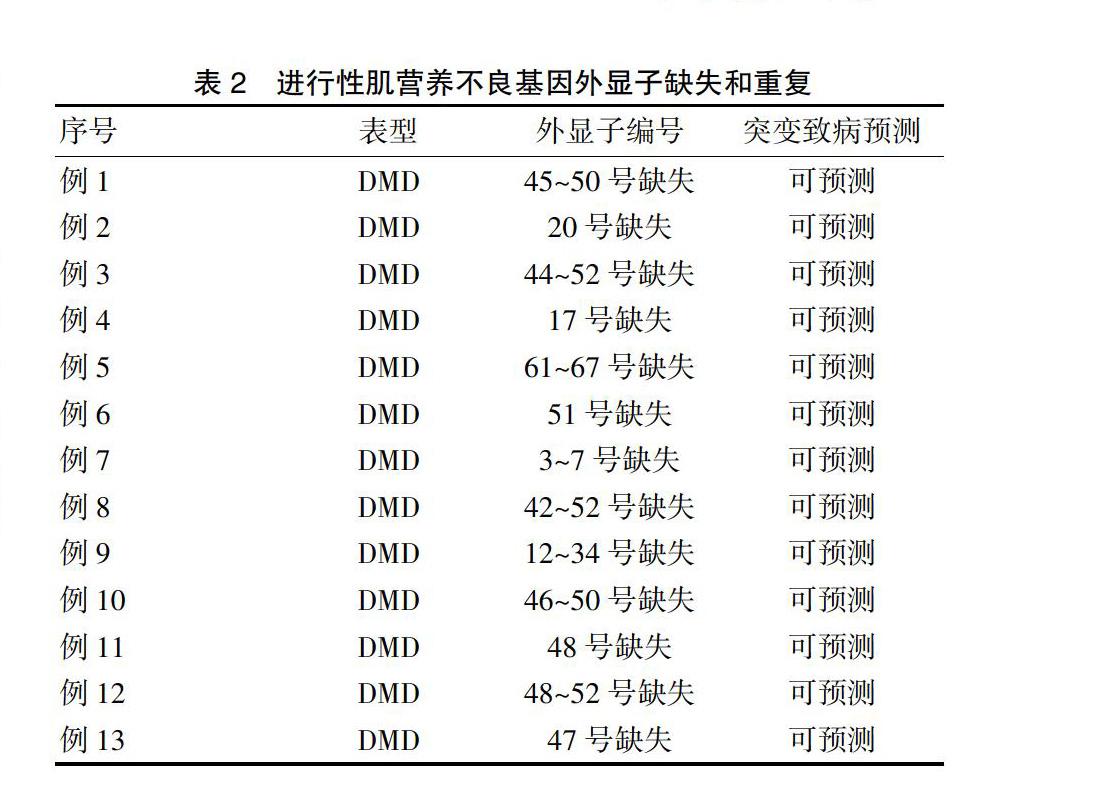
2.2.3 基因檢測(cè)及肌肉活檢 ?30例(90.9%)根據(jù)MLPA技術(shù)檢測(cè)出其基因的外顯子缺失或者重復(fù),13例有記錄明確突變的位點(diǎn),其余部分患者資料遺失。其中3例(9.1%)未檢測(cè)出異常結(jié)果,2例(6.1%)進(jìn)一步行NGS提示異常,有3例(9.1%)行肌肉活檢明確有肌細(xì)胞壞死、肌肉組織纖維變性。見表2。
2.2.4 心臟彩超及心電圖 ?根據(jù)患者情況選擇性進(jìn)行心臟彩超及心電圖檢查,15例(45.5%)行心臟彩超,其中1例提示肺動(dòng)脈內(nèi)徑輕度增寬,1例提示左右房?jī)?nèi)徑稍增寬,1例左室前壁運(yùn)動(dòng)稍欠協(xié)調(diào),左室射血分?jǐn)?shù)稍下降(55%~57%)。17例行常規(guī)心電圖,其中7例提示竇性心律不齊,4例提示左心室高電壓,1例有竇房結(jié)-房下部游走性心律,1例有短陣加速性房性自主心律,1例有不完全右束支傳導(dǎo)阻滯,V1~V2導(dǎo)聯(lián)可疑異常Q波。
2.3 治療
33例均予輔酶Q10或果糖二磷酸營(yíng)養(yǎng)肌肉治療,4例予小劑量激素治療(0.5 mg/kg)[6],但療效欠佳,病情仍進(jìn)展。
2.4 隨訪
隨訪27例,6例失訪,隨訪率達(dá)81%,隨訪時(shí)間3個(gè)月~6年不等,12例已不能行走,需臥床、坐輪椅,且其中5例已有肌肉萎縮、關(guān)節(jié)變形。8例出現(xiàn)行走困難,7例尚只有酶譜異常。
3 討論
進(jìn)行性肌營(yíng)養(yǎng)不良是由抗肌萎縮蛋白基因突變引起,也稱為抗肌萎縮蛋白病。由于肌纖維變性是主要的病理過(guò)程,故肌無(wú)力是主要的癥狀[7]。其患病率在活產(chǎn)男嬰中約為1/5000,因近年隨著基因檢測(cè)的技術(shù)逐漸發(fā)展,該病的檢出率升高[8]。
本研究納入33例在我院確診的進(jìn)行性肌營(yíng)養(yǎng)不良患兒,多為DMD型,且以男性為主,符合X連鎖隱性遺傳規(guī)律。2例患兒有家族史,同樣為男性發(fā)病。該研究中有3例女性發(fā)病,可能其發(fā)病機(jī)制上存在X染色體偏移失活[9],女性患兒臨床癥狀輕微,病情相對(duì)進(jìn)展緩慢。發(fā)病年齡也具有一定特點(diǎn),11例出現(xiàn)典型癥狀及體征的孩子,發(fā)病年齡多在3~4歲,22例發(fā)病時(shí)無(wú)肢體無(wú)力,行走困難,步態(tài)異常等表現(xiàn),但隨訪患兒后,發(fā)現(xiàn)其部分患兒病情進(jìn)展,逐漸不能行走。
33例患兒均發(fā)現(xiàn)明顯的谷丙轉(zhuǎn)氨酶、谷草轉(zhuǎn)氨酶及肌酸肌酶升高,進(jìn)行分析后發(fā)現(xiàn)肝臟酶譜升高在2~3倍以上,肌酸激酶及其同工酶升高在10倍以上,因Ckmb/CK小于10,故考慮肌肉來(lái)源的病變?yōu)橹鳌Q芯堪l(fā)現(xiàn)在疾病未出現(xiàn)任何臨床癥狀前,患者就有酶譜升高。本研究中大部分患兒心臟彩超未見明顯異常,少數(shù)患兒有影響左室的運(yùn)動(dòng)及射血分?jǐn)?shù)下降,部分患兒心電圖中有左心室高電壓,不完全右束支傳導(dǎo)阻滯等,但沒有特異性改變。考慮隨著進(jìn)行性肌營(yíng)養(yǎng)不良的過(guò)程發(fā)展,部分患者出現(xiàn)心電圖異常,因心肌纖維化有可能導(dǎo)致心電圖的特征性變化,也有可能伴傳導(dǎo)障礙,尤其心房?jī)?nèi)和心房間傳導(dǎo),從而導(dǎo)致多種心律失常(以室上性為主)[10]。此外,心臟線粒體功能障礙會(huì)加重,這也是其心臟病變的原因[11]。
目前研究已發(fā)現(xiàn)DMD的基因位于Xp21.1,在其中橫跨大約230萬(wàn)個(gè)堿基,蛋白產(chǎn)量也非常大,分子量為427 kD。是人類已知的最大的基因,有79個(gè)外顯子[12],所以遺傳診斷是治療的基礎(chǔ)。本文有30例患者先進(jìn)行MLPA技術(shù)檢測(cè),排查DMD基因缺失或重復(fù),2例再行NGS測(cè)序應(yīng)排除MLPA陰性樣品。同時(shí),聚合酶鏈反應(yīng)(PCR)用于檢測(cè)單外顯子缺失,以排除點(diǎn)突變引起的MLPA中的假陽(yáng)性[13]。發(fā)現(xiàn)其DMD基因缺失部位主要在2個(gè)區(qū)段,以45~54號(hào)及2~20號(hào)最為常見[14],但臨床表現(xiàn)與缺失或者重復(fù)的片段部位無(wú)太大相關(guān)性,與表達(dá)的閱讀框有很明顯的相關(guān),若閱讀框的表達(dá)受影響,則臨床的表現(xiàn)及體征明顯。肌肉活檢雖是診斷的金標(biāo)準(zhǔn),但不是其適應(yīng)證,且因其為創(chuàng)傷性檢查,故臨床的應(yīng)用相對(duì)其他兩種少[15]。
目前針對(duì)進(jìn)行性肌營(yíng)養(yǎng)不良沒有特效藥,本研究納入的患兒均使用輔酶Q10或果糖二磷酸片營(yíng)養(yǎng)肌肉,減緩病變,但病情仍進(jìn)展。隨訪的部分患兒使用小劑量激素治療,但效果欠佳。目前新的治療方法仍在探索中,其中有針對(duì)無(wú)意義的突變治療,是利用一種口服的生物小分子誘導(dǎo)核糖體無(wú)意義突變并部分恢復(fù)肌營(yíng)養(yǎng)不良蛋白的產(chǎn)生。此外有框外缺失的反義寡核苷酸[16]、AAV基因療法等[17],但療效尚不明確。
目前納入的病例數(shù)有限,隨訪時(shí)間未達(dá)到終點(diǎn),尚需要更多的臨床研究進(jìn)一步探討。
進(jìn)行性肌營(yíng)養(yǎng)不良有其典型的表型及基因突變特征,目前對(duì)于其治療方案尚需要更多研究探討,本病可以通過(guò)基因檢測(cè)或者肌肉活檢確診,對(duì)二胎的產(chǎn)前診斷有益[18-19],進(jìn)而降低該病的發(fā)生率及死亡率。
[參考文獻(xiàn)]
[1] Bushby K,F(xiàn)inkel R,Birnkrant DJ,et al. Diagnosis and management of Duchenne muscular dystrophy,part 1:Diagnosis,and pharmacological and psychosocial man-agement[J].Lancet Neurology,2010,9(1):77-93.
[2] Kwon JM,Abdel-Hamid HZ,Al-Zaidy SA,et al.Clinical follow-up for duchenne muscular dystrophy newborn screening:A proposal[J]. Muscle Nerve,2016,54(2):186-191.
[3] Muntoni F,Torelli S,F(xiàn)erlini A,et al. Dystrophin and mutations:One gene,several proteins,multiple phenotypes[J].Lancet Neurology,2003,2(12):731-740.
[4] 季蘇瓊,李悅,王瓊,等.杜氏肌營(yíng)養(yǎng)不良癥的臨床表現(xiàn)與基因表型的關(guān)聯(lián)[J]. 神經(jīng)損傷與功能重建,2016,11(1):31-34,45.
[5] Mirski KT,Crawford TO.Motor and cognitive delay in Duchenne muscular dystro-phy:Implication for early diagnosis[J].J Pediatr,2014,165(5):1008.
[6] 李婷婷,鄒麗萍.糖皮質(zhì)激素治療Duchenne肌營(yíng)養(yǎng)不良的研究進(jìn)展[J].中華神經(jīng)醫(yī)學(xué)雜志,2012,11(11):1182-1185.
[7] Okubo M,Goto K,Komaki H,et al.Comprehensive analysis for genetic diagnosis of dystrophinopathies in Japan[J].Orphanet Journal of Rare Diseases,2017,12(1):149.
[8] Parsons EP,Clarke AJ,Hood K,et al. Newborn screening for Duchenne musc-ular dystrophy:A psychosocial study[J].Archives of Disease in Childhood-Fetal and Neonatal Edition,2002,86(2):F91.
[9] 吳海榮,馬祎楠,戚豫,等.攜帶DMD/BMD基因變異女性的X染色體失活研究[J].第九屆全國(guó)遺傳病診斷與產(chǎn)前診斷學(xué)術(shù)交流會(huì)暨產(chǎn)前診斷和醫(yī)學(xué)遺傳學(xué)新技術(shù)研討會(huì)論文集,2014.
[10] Yoo WH,Cho MJ,Chun P,et al.The evolution of electrocardiographic changes in patients with duchenne muscular dystrophies[J]. Korean Journal of Pediatrics,2017,60(6):196-201.
[11] Gabriella Esposito,Antonella Carsana.Metabolic alterations in cardiomyocytes of patients with duchenne and becker muscular dystrophies[J].Clin Med,2019,8(12):21-51.
[12] Zhong J,Xu T,Chen G,et al.Genetic analysis of the dystrophin gene in children with duchenne and becker muscular dystrophies:Genetic analysis in DMD/BMD[J].Muscle & Nerve,2016,56(1):117-121.
[13] 白瑩,李雙,宗亞楠.杜氏/貝氏肌營(yíng)養(yǎng)不良癥433個(gè)家系的基因突變分析[J].中華醫(yī)學(xué)雜志,2016,96(16):1261-1269.
[14] 賀影忠,韓鳳,王紀(jì)文,等.Duchenne型進(jìn)行性肌營(yíng)養(yǎng)不良臨床和基因變異97例分析[J].中國(guó)實(shí)用兒科雜志,2019,34(1):38-41.
[15] Taylor LE,Kaminoh YJ,Rodesch CK,et al.Flanigan quantification of dystrophin immunofluorescence in dystrophinopathy muscle specimens[J].Neuropathology and Applied Neurobiology,2012,38:591-601.
[16] Nakamura,Akinori.Moving towards successful exon-skipping therapy for duchenne muscular dystrophy[J].Journal of Human Genetics,2017,128:54-56.
[17] Eugenio Mercuri,Carsten G B?觟nnemann,F(xiàn)rancesco Muntoni,et al.Muscular dystrophies[J].Lancet,2019,394:2025-2038.
[18] 趙煒,姜楠,李朔,等.假性肥大型肌營(yíng)養(yǎng)不良癥的遺傳學(xué)分析及產(chǎn)前診斷[J].中華婦產(chǎn)科雜志,2019,54(4):226-231.
[19] 李煥錚,徐晨陽(yáng),毛義建,等.50個(gè)假肥大型肌營(yíng)養(yǎng)不良家系的基因突變檢測(cè)及產(chǎn)前診斷[J].中華醫(yī)學(xué)遺傳學(xué)雜志,2018,35(2):169-174.
(收稿日期:2020-02-18)
猜你喜歡
少兒美術(shù)·書法版(2021年12期)2021-10-24 02:50:16
少兒美術(shù)·書法版(2021年9期)2021-10-20 06:35:28
少兒美術(shù)·書法版(2021年7期)2021-10-20 06:29:16
少兒美術(shù)·書法版(2021年11期)2021-10-20 06:23:28
少兒美術(shù)·書法版(2021年10期)2021-10-20 06:14:04
少兒美術(shù)·書法版(2021年8期)2021-10-20 06:08:10
少兒美術(shù)(2019年8期)2019-12-14 08:07:00
少兒美術(shù)(2019年3期)2019-12-14 08:02:56
雜文選刊(2016年7期)2016-08-02 08:39:56
小天使·一年級(jí)語(yǔ)數(shù)英綜合(2016年6期)2016-05-14 12:21:05

- 中國(guó)現(xiàn)代醫(yī)生的其它文章
- 綠原酸抑制TLR4/NF-κB通路誘導(dǎo)巨噬細(xì)胞源性泡沫細(xì)胞凋亡的進(jìn)展分析
- 快速康復(fù)理念在神經(jīng)根型頸椎病經(jīng)皮頸椎間盤摘除術(shù)圍手術(shù)期的應(yīng)用
- 釘釘平臺(tái)在改進(jìn)婦科護(hù)士在職培訓(xùn)管理中的應(yīng)用研究
- 中藥涂擦配合穴位按摩治療腦卒中肩手綜合征Ⅰ期的療效觀察
- NOSESⅣ型改良手術(shù)聯(lián)合加速康復(fù)理念在乙狀結(jié)腸癌治療中的應(yīng)用1例
- 柴芍六君湯治療肝郁脾虛型潰瘍性結(jié)腸炎的療效觀察