非小細胞肺癌中靶向RET、FGFR融合基因的研究進展
2020-08-26 10:41:48鐘煒祥韋晰鳳
實用醫學雜志 2020年15期
鐘煒祥 韋晰鳳
1贛南醫學院第一附屬醫院心胸外科(江西贛州341000);2贛州市婦幼保健院(江西贛州341000)
肺癌已成為我國癌癥相關發病率和病死率第1 的惡性腫瘤,80%~85%的肺癌屬于非小細胞肺癌(non-small cell lung cancer,NSCLC),后者主要包括肺腺癌(lung adenocarinoma,LADC)和肺鱗狀細胞 癌(lung squamous cell carcinoma,LSQCC)。NSCLC 的治療目前已跨入個體化靶向精準治療的時代,以表皮生長因子受體(EGFR)和間變性淋巴瘤激酶(ALK)、C-ros 原癌基因1-受體酪氨酸激酶(ROS1)融合基因作為分子靶點的靶向藥物(如吉非替尼[1]、厄洛替尼[2]、克唑替尼[3]等)針對性強、療效可靠、不良反應輕,極大地改善了患者的預后。融合基因變異也因此成為了當前肺癌分子靶向治療研究的熱點。
轉染時發生重排(rearranged during transfection,RET)融合基因是在NSCLC 患者中繼EGFR、ALK等基因之后發現的新型突變基因,研究[4]表明RET 融合基因可能是EGFR、ALK、ROS1 均未突變肺癌患者的主要發病途徑,抑或是此類NSCLC患者致病的新靶點。目前,高選擇性RET 抑制劑BLU-667 和LOXO-292 均取得了重大突破,值得期待。
表皮生長因子受體-酪氨酸激酶抑制劑(EGFR-TKIs)的出現使LADC 的靶向治療成為現實。但由于LSQCC 的EGFR 突變和ALK、ROS1 重排率低,除了傳統的手術、化療、放療外,目前仍缺乏有效的靶向治療。因此,積極致力于LSQCC 的相關驅動基因的研究,找尋潛在的治療靶點,以實現LSQCC 患者的個體化靶向精準治療。成纖維細胞生長因子受體(fibroblast growth factor receptor,FGFR)作為LSQCC 中突變頻率最高的酪氨酸激酶家族基因[5],FGFR 基因融合后,細胞的FGFR 激酶區活性增強[6],進而誘發腫瘤的產生和發展。研究[7]亦表明,FGFR 抑制劑可以抑制FGFR3-TACC3融合基因的酪氨酸激酶活性。因此,FGFR 融合基因,特別是FGFR3-TACC3 已成為LSQCC 切實可行的治療靶點,有望LSQCC 靶向治療的新方向。
2019年ASCO年會上公布了高選擇性RET 抑制劑BLU-667 的最新研究成果和FGFR-TACC 融合基因圖譜,受到廣泛關注。本文圍繞RET、FGFR基因的生物學特性,NSCLC 中RET、FGFR 基因融合的臨床病理特征和RET、FGFR 抑制劑在NSCLC靶向治療中的應用前景進行綜述。
1 RET 融合基因
1.1 RET 基因的生物學特性RET 基因是在NSCLC 患者中繼EGFR、ALK 等基因之后發現的新型突變基因,位于第10 號染色體(10q 11.2),長約60 kb,含21 個外顯子,編碼RET 蛋白,后者是由1 143 個跨膜氨基酸殘基構成的蛋白聚合體,屬于酪氨酸激酶(TK)受體。作為RET 蛋白的配體,膠質細胞衍生的神經營養因子家族(包括GDNF、NINneuturin、ART artimin、PSP persephin)通過糖基化的磷脂酰錨定蛋白與RET 蛋白結合,使后者的酪氨酸激酶區出現磷酸化,進而激活下游包括磷脂酶C、磷脂酰肌醇3-激酶和RAS/有絲分裂原激活蛋白激酶途徑等多條信號轉導通路[8],最終調節細胞生存和誘導細胞增生。
RET 基因的融合突變是通過第10 號染色體上的臂間倒位并致RET 基因斷裂,結合其他基因發生重組,最終形成新的融合基因,后者可直接導致酪氨酸酶自身活化,自發向細胞內傳遞信息,最終誘使細胞增殖過度并生成腫瘤。
1.2 NSCLC 中RET 基因融合的臨床病理特征RET 融合基因于2012年在NSCLC 中首次發現,發生概率維持在1%~2%[9-11]。CONG 等[9]通過檢索The PubMed,SpringerLink 等數據庫入組了13 篇論文,納入8 859 個病例,檢測出169例RET 融合基因,陽性表達率為1.91%,其中121例為KIF5BRET,后者多為女性且相對年輕(<60 歲)。同時指出RET 基因融合在非吸煙LADC 患者中常見。自發現以來,RET 融合基因被認為與其他突變基因相互排斥,但越來越多研究[12-13]證實RET 融合基因可與EGFR、MAP2K1、CTNNB1、AKT1、TP53、SETD2 及MET 擴增等基因變異共存,LU 等[13]還指出合并TP53 突變的RET 基因融合NSCLC 患者OS更短。此外,有研究表明RET 基因融合可能是導致一代EGFR-TIKs[14]和Osimertinib[15](三代EGFRTIKs)獲得性耐藥的機制之一。
RET 基因在NSCLC 能與多種異源上游伴侶基因發生融合[16],其中KIF5B-RET 型最常見,其次為CCDC6-,其余為RET少見融合類型。目前在NSCLC中至少發現了23 種少見的RET 融合類型[17-27]。
1.3 RET 抑制劑在NSCLC 靶向治療中的應用前景目前尚無特異性的RET 抑制劑上市,但一些多靶點抑制劑(MKIs)可以覆蓋RET 基因融合靶向治療。根據MKIs 的作用機制不同分為:可與活化的RET 激酶結合的Ⅰ型抑制劑(vandetanib 和sunitinib)和與非活化的RET 激酶結合的Ⅱ型抑制劑(cabozantinib、sorafenib、ponatinib 等)。
表1列出了單一MKI 在晚期預處理的RET 基因融合NSCLC 中的Ⅱ期單臂臨床試驗,Sorafenib研究中入組3例患者,其中1例穩定(SD),2例進展(PD),沒有表現出明顯的療效;Cabozantinib、Vandetanib、Lenvatinib 在RET 基因融合晚期NSCLC患者中的客觀緩解率(ORR)為16%~53%,中位無進展生存期(mPFS)為(4.5~7.3)個月,均劣于針對EGFR/ALK/ROS1 驅動基因抑制劑(在EGFR/ALK/ROS1 陽性的NSCLC 患者中[28-30],其對應抑制劑治療的ORR分別為56%~85%、65%~90%、65%~85%,mPFS 分別為(9.2~13.7)、(8~11)、(9.3~19.3)個月。其中,Vandetanib 的兩個Ⅱ期臨床試驗來自日本和韓國,發現CCCD6-RET 病例接受Vandetanib 治療可獲得比KIF5B-RET 更高的ORR。2017年,一項全球多中心的回顧性研究[16]分析了RET 基因融合晚期NSCLC 患者接受多種MKIs 治療的效果,結果顯示Cabozantinib、Vandetanib、Sunitinib 治療組的ORR 分別為37%、18%及22%,mPFS分別為3.6、2.9、2.2 個月,mOS 分別為4.9、10.2、6.8 個月;而在Sorafenib、Alectinib、Ponatinib、Regorafenib 治療組均未見完全或部分緩解的病例。這些研究結果表明目前常見的靶向RET 基因的MKIs 療效有限,考慮可能同時存在MDM2 擴增[31]或RET S904F 突變[32]或守門突變V804M/L[33]等因素,進而導致MKIs 獲得性耐藥,有待進一步考證。此外,Alectinib(NCT03131206 和UMIN000020628),sunitinib(NCT01829217)、ponatinib(NCT01813734)及Apatinib(NCT02540824)等MKIs 正在進行Ⅰ~Ⅱ期臨床試驗,期待它們最終的研究結果。
新型高選擇性RET 抑制劑BLU-667 和LOXO-292 應運而生并取得了重大突破。目前針對BLU-667 和LOXO-292 正在進行兩個Ⅰ期臨床試驗:ARROW(NCT03037385)和LIBRETTO-001(NCT03 157128),2018年研究者分別在ASCO 和AACR年會上公布了前期結果:LOXO-292 的ORR 為68%,BLU-667 的ORR 為50%[38-39]。在2019 ASCO年會上,研究者更新了ARROW 研究中BLU-667 治療RET 融合陽性NSCLC 患者的進一步研究[40]結果,在48例有可測量病灶且至少進行過一次隨訪評估的患者中,ORR 為58%(1例完全緩解(CR),27例部分緩解(PR),18例SD,2例PD,疾病控制率(DCR)為96%(46/48);在既往接受過鉑類化療的35例患者中,其ORR 高達60%(1例CR,20例PR,14例SD),DCR 為100%。Ⅰ期臨床試驗結果顯示,LOXO-292和BLU-667在RET 融合陽性晚期NSCLC患者中具有強效、持久和廣泛的抗腫瘤活性,除了常見的KIF5B-RET,對各種潛在的RET 耐藥變異如CCDC6-RET 融合、RET 點突變C634W、M918T、V804L/M等均有很好的抑制[39,41]。二者入腦能力很強,對顱腦轉移瘤有效,而且耐受性良好,均獲得FDA“突破性療法”認定,是目前精準靶向治療的選擇。
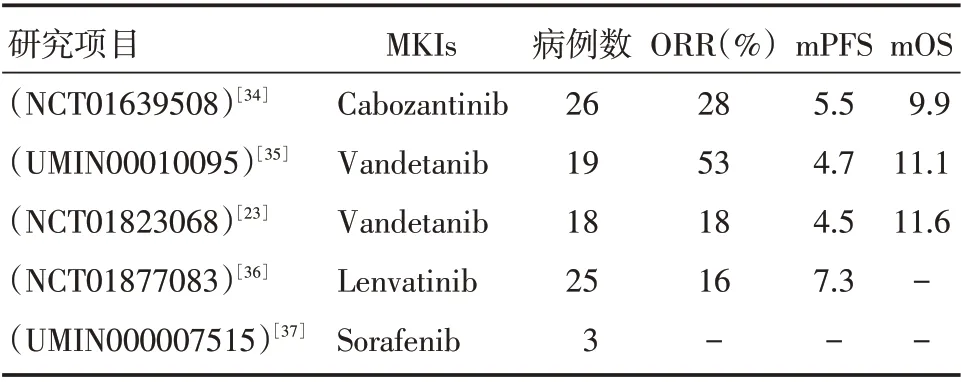
表1 晚期預處理的RET 重排的NSCLC 中單一MKI 的Ⅱ期臨床試驗Tab.1 Ⅱphase clinical trials of single MKI in advanced RET-positive NSCLC pre-treated
2 FGFR 融合基因
2.1 FGFR 基因的生物學特性FGFR 屬于一類跨膜酪氨酸激酶受體家族(由胞外區,跨膜區及具有TK 的胞內區構成),為單鏈糖蛋白分子[相對分子質量為(1.1~1.5)×105],一共包括FGFR1、FGFR2、FGFR3、FGFR4 及FGFRL1(其中,FGFR1-4 具有酪氨酸激酶區域,FGFRL1 無蛋白酪氨酸激酶區域)。成纖維細胞生長因子(FGFs)是FGFRs 的配體,目前已發現23 個FGFs(FGF1-FGF23)。FGFRs依賴于細胞表面的核心結合位點HSPG 結合FGFs形成二聚體,FGFR 胞內區TK 殘基接著發生磷酸化,進而激活Ras-Raf-Mapk、P13K-Akt、Stats 和PLCr 四條主要下游信號通路產生生物學效應[42],導致腫瘤的發生和轉移瘤的增長。
2.2 NSCLC 中FGFR 融合基因的臨床病理特征縱覽近5年有關NSCLC 中FGFR 基因融合的相關文獻[43-46],FGFR1-4 基因融合在NSCLC 中的發生概率維持在0.1%~1.3%,至少發現20 種不同的FGFR 融合形式,其中FGFR3-TACC3 是最常見的融合類型。蔡修宇教授在2019年ASCO年會上報道了一項亞洲人群NSCLC 中FGFR-TACC 融合基因融合譜的研究,在2 743例NSCLC 中發現FGFRTACC 融合16例(0.58%),其中1例FGFR1-TACC1,3例FGFR2-TACC2,12例FGFR3-TACC3 融合。蔡修宇教授認為FGFR-TACC融合作為NSCLC 的一個亞型,可能對FGFR 抑制劑(如AZD4547 等)具有潛在的臨床獲益可能。此外,王磊等[45]指出,FGFR融合基因在LSQCC 組陽性率高達3.4%,好發于腫瘤最大徑>3 cm 的男性吸煙患者。此與以往靶基因突變多好發于不吸煙女性LADC 患者不同,為LSQCC 的靶向治療帶來了新的思路。
2.3 FGFR 抑制劑在NSCLC 靶向治療中的應用前景目前亦無選擇性的FGFR 抑制劑上市,針對FGFR 基因為靶點正處在試驗研究的的藥物主要包括3 類:選擇性FGFR 抑制劑、FGF-FGFR 結合抑制劑(FP-1039/GSK3052230)和非選擇性多靶點FGFR 抑制劑(Nintedanib、Dovitinib 等),其中選擇性FGFR 抑制劑主要包括AZD4547(FGFR1-3 抑制劑)、BGJ398(FGFR1-3 抑制劑,對FGFR1 的抑制活性最強)、LY2874455(FGFR1-4 抑制劑)和JNJ-42756493(即Erdafitinib,針對FGFR1-4 的多靶點TKI)。
表2列出了FGFR 抑制劑在晚期NSCLC 中進行的Ⅰ~Ⅲ期臨床試驗,研究結果表明AZD4547、BJG398、Nintedanib 及Divotinib 均對FGFR 變異(特別是FGFR1 擴增)NSCLC(特別是LSQCC)表現出一定的抗腫瘤活性,ORR 維持在8%~11.5%。其中,在針對Nintedanib 治療晚期NSCLC 多中心隨機對照Ⅲ期臨床試驗LUME-lung1 中,一線治療后9 個月內的LADC 患者采用Nintedanib 聯合多西他賽治療方案的OS 明顯比對照組更長,研究者指出Nintedanib 聯合多西他賽治療方案可作為含鉑雙藥一線化療后進展的晚期NSCLC,特別是LADC患者的一個行之有效的二線治療方案選擇。此外,針對FGFR 抑制劑的臨床試驗如(AZD454,Ⅱ~Ⅲ期)NCT02154490、(BGJ398,Ⅰ期)NCT01697605、(Erdafitinib,Ⅱ期)NCT02699606、(FP-1039,Ⅰ期)NCT01604863;(FP-1039,Ib 期)NCT01868022)、(Dovitinib,Ⅱ期)NCT01676714 等正在進行中,結果值得期待。
作為FGFR基因常見的變異類型,FGFR基因融合后,細胞的FGFR激酶區活性增強[6],HARSHNIRA等[51]對比了FGFR 抑制劑AZD4547 和JNJ42756493對NIH3T3 細胞系中FGFR-TACC3 融合基因的治療效果,發現1 000 nmol/L的JNJ42756493對于這種細胞的增殖具有較強的抑制作用;CAPELLETTI 等[52]利用濃度遞增的BGJ398、Ponatinib 處理含有FGFRTACC3融合的Ba/F3細胞,均能有效抑制后者生長;PERERA 等[53]發現Erdafitinib 在體內的抗腫瘤活性在FGFR 基因融合模型中最突出,高于FGFR 基因擴增模型。目前對于FGFR基因融合的靶向研究絕大多數處在臨床前試驗階段,現有的研究表明FGFR 抑制劑可以抑制FGFR3-TACC3 融合基因的酪氨酸激酶活性[7]。因此,FGFR 融合基因,特別是FGFR3-TACC3 融合類型確實是可行的治療靶標,且可能相較其他FGFR 突變具有更靈敏的FGFR 靶向治療位點。

表2 針對FGFR 抑制劑治療NSCLC 的Ⅰ~Ⅲ期臨床試驗Tab.2 Ⅰ~Ⅲphase clinical trials of FGFR inhibitors in NSCLC
3 NSCLC 中融合基因的檢測方法
迄今為止,尚未發現檢測融合基因的特異性方法,常用的檢測方法包括熒光原位雜交(FISH)、免疫組化(IHC)、逆轉錄聚合酶鏈反應(RT-PCR),其中FISH 已獲FDA 批準用于在NSCLC 患者中檢測ALK 融合。表3列舉了基因融合或融合蛋白的常用分子診斷各種方法的優缺點。
此外,二代測序(next generation sequencing,NGS)對于近年來NSCLC 的分子診斷具有革命性的意義,具有高通量、自動化、低成本的特征,能夠一次性同時檢測EGFR、ALK、ROS1、RET 及FGFR等多個基因變異,同時鑒別出已知和未知的突變類型,檢測敏感性高于一代測序,可以檢測基因1%的低頻突變。

表3 免疫組織化學、熒光原位雜交和即時熒光定量PCR(RT-PCR)檢測方法的比較Tab.3 Comparison of detection methods in immunohistochemistry,fluorescence in situ hybridization and real-time fluorescence quantitative PCR(RT-PCR)
以上檢測方法,特別是NGS 為臨床試驗研究提供技術保障。但由于NGS 技術的復雜性,NGS質量體系的標準化、規范化成為目前NGS 在臨床常規開展的瓶頸,而且NGS 的收費還比較高,暫不推薦臨床常規使用NGS 檢測肺癌各靶向基因。
針對EGFR、ALK、ROS1 均未突變NSCLC 患者的靶向治療是目前臨床亟待解決的重點問題,深入研究RET、FGFR 等少見突變基因在NSCLC 中的靶向作用機制,是未來RET、FGFR 等少見突變基因變異的NSCLC 患者靶向治療的新思路,有利于EGFR、ALK 基因抑制劑耐藥后再次靶向治療的靶位尋找。隨著個體化精準治療模式的廣泛應用和NGS 技術的快速發展,對NSCLC 患者進行驅動基因檢測是精準藥物治療的基礎,常規檢測RET、FGFR 等少見突變基因在NSCLC 的診治中或將成為臨床診斷共識,指導肺癌的分子靶向治療,篩選出獲益的靶向治療人群,實現個體化精準治療。期待L0XO-292 和BLU-667Ⅰ期臨床試驗的最終結果,盡快進行Ⅱ、Ⅲ期臨床試驗并最終獲批應用于臨床,給RET 重排NSCLC 患者帶來積極的生存獲益,彌補RET 融合變異NSCLC 現存治療的空白;期待隨著對FGFR 融合基因靶向作用機制的深入研究并開發出更多高選擇性FGFR 抑制劑,從而使LSDCC 的靶向治療也能像LADC 一樣占據著舉足輕重的作用,真正地為NSCLC 患者的個體化靶向治療帶來福音。
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
今日農業(2021年19期)2022-01-12 06:16:36
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年11期)2021-08-22 03:15:44
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
無線電工程(2020年11期)2020-10-29 01:25:46
