普魯蘭酶基因在畢赤酵母中的表達研究
2020-08-27 12:58:25王顥霖
河南農業·教育版 2020年7期
王顥霖
關鍵詞:普魯蘭酶;克隆;畢赤酵母;誘導表達
在淀粉的加工行業中,高壓酸解法作為葡萄糖生產的老工藝具有需要設備少、耗時短的優點,但其副產物較多,耗費的酸和堿的量較高,而且為了去除苦味必須采用嚴格控制溫度、緩慢結晶的辦法,需要淀粉加工工廠投入較高的人力資源和設備成本;如果運用a-淀粉酶作用于較長的淀粉分子使其斷裂成小分子,使高濃度的淀粉糊在高溫下糊化液化,再運用糖化型淀粉酶,就可以高效地得到目的產物。此法具有生產率高、成本低、副產物少、結晶快等優點,對于生產實踐具有很大的意義。
普魯蘭酶最早是在肺炎克雷伯氏菌中被發現,并且被發現可以水解支鏈淀粉中的a-1,6糖苷鍵,不僅能夠極大提高淀粉的水解效率,還能得到具有高特異性的產物,對于淀粉的糖化步驟十分重要。自然界中的天然酶一般都具有含量低、提取較難、不易制備的弊端,通常在自然界中篩選特定性質酶的效率很低。而研究酶在工程菌株中的克隆與表達,選育出更好的產酶菌株,對于獲得更高酶活性,更高表達量的性能優良的酶具有非常重要的意義。
各項研究表明,普魯蘭酶可被許多菌種表達,如緩和鏈球菌,大腸桿菌,耐熱產硫梭菌,枯草芽孢桿菌,酵母菌等等。在原核表達系統中,大腸桿菌表達系統作為研究較為深入的表達系統,在1984年最早成功異源表達了普魯蘭酶,但是其翻譯后修飾的過程比較簡單,表達量也很低。經過科研人員的不斷努力,對表達宿主大腸桿菌進行優化和對發酵條件進行改善,生產出的酶活力曾經高達502U/mL,具有很強的工業應用前景。但是其高密度發酵時的溫度一般是低于37℃的,不利于細菌的快速生長;枯草芽孢桿菌也具有清晰的遺傳背景和生理特性,未經優化的情況下,表達普魯蘭酶的活力可達到10.94U/mL。兩者都具有一定的工業生產普魯蘭酶的潛力。
真核生物Pichia pastoffs最早是在1987年被Cregg研究將其作為宿主成功的進行了外源蛋白的表達實驗,被發現其在外源表達方面所具有的潛力。隨著人們對Piehia pas-toffs研究的深入,發現Pichiapastoffs在外源表達方面的獨特優勢,與其他表達系統比較起來的優點大體上為:對細胞沒有毒害,是一種安全的表達宿主。表達的量通常較高,可以獲得大量的目的蛋白,部分蛋白的表達量高至12g/L。不但可以用于分泌型表達,還可以用于細胞內表達。表達出的蛋白易于分離純化。某些在細菌系統中表達不好的真核基因,在酵母系統中表達情況良好。能夠在廉價的培養基上生長,十分節省實驗成本,方便用于外源基因的操作。作為真核生物,其轉錄翻譯后加工、外分泌、翻譯后修飾以及糖基化修飾的性能良好,可以使表達產物更加接近于天然蛋白質,甚至與天然蛋白質一樣。利用高表達的啟動子MOX、AOX、LAC4等,外源基因可同源重組到Pichia pastoris的基因組中進行穩定復制,有利于外源基因的表達。曾用于生產SCP,有著清晰的遺傳背景和良好的發酵基礎。細胞生長速度快,適用于高密度發酵能夠移去起始Met,使重組蛋白在用于臨床應用時不引起免疫反應。因此,對醫療行業起到很大的貢獻。
實驗前期將來自于本實驗室的普魯蘭酶基因在大腸桿菌BL21 DE3中進行表達,其表達量非常低。換用能緩解大腸桿菌稀有密碼子問題大腸桿菌Rossta DE3也未能明顯提高蛋白的表達量。
本實驗旨在通過以更換表達宿主的方式提高蛋白的表達量。即將目的基因同源重組入PichiapastorisGSll5,對重組菌進行誘導表達,分析其蛋白產率。
一、材料與方法
(一)材料
1、菌株和質粒。含有Thermogota thermarum中獲取的普魯蘭酶基因的重組質粒pET21a-TT由河南農業大學推廣樓酶工程504實驗室構建和保存,畢赤酵母(Piehia pasto-ris P.pastoris)GSll5和表達載體pPIC3.5K由河南農業大學酶工程實驗室保存。
2、工具酶和主要試劑。山梨醇(D-Sorbit01),瓊脂(Agar Powder),酵母提取物(Yeast Extract),質粒小提試劑盒,PCR產物回收試劑盒,膠回收DNA試劑盒,椰油醇硫酸鈉(SDS)胰蛋白胨(Ttyton),瓊脂糖(Agarose),三羥基甲基氨基甲烷(riffs),氨芐青霉素(Ampicillin),甘氨酸(Glycine),三羥甲基氨基甲烷(Tris),乙二胺四乙酸(EDTA)等。甲醇,異丙醇等普通試劑均為分析純試劑。各種工具酶購自SMOBIL公司。設計引物如下:
TF-EcoRI-F:CCGGAATFCGCCACCATGAAAAGGCTAC-337TACTCATCGTCATAACTTGTACAG
Tr-NotI-R:ATAAGAATGCGGCCGCTCAGTGGTGGTGG-TGGTGG
5"AOX GACTGGTTCCAATYGACAAGC;
3"AOX GCAAATGGCATTCTGACATCC
引物交由蘇州金唯智生物科技有限公司合成。目的基因和載體測序以及轉化子測序由鄭州何澤公司完成。
(二)方法
1、含目的基因載體的驗證。從-80℃冰箱取出10%甘油菌保存的重組菌劃線于帶氨芐抗性的LB平板,在恒溫培養箱37℃培養活化12~16h。挑取單菌落于5mL帶氨芐抗性LB培養試管中,放置于搖床37℃,220 rpm培養12~16h。提取質粒。測定濃度并進行瓊脂糖凝膠電泳,將位置正確的質粒送去測序公司進行測序。
2、重組質粒的構建。使用引物TT-EcoRI-F與TT-No.tI-R擴增目的基因片段。按如下體系將樣品加入PCR管:ddH20 38 txL;TT-EcoRI-F 2.5μL;TY-Nod-R 2.5μL;pET21a-TY 1μL;10xQ5聚合酶緩沖液5μL;Q5聚合酶1μL。設置程序:98℃預變性2 min;98~C變性15 s,66℃退火15 s,72℃延伸2 min,30個循環;72℃再延伸2 min;16℃保存,完成后取3tμL瓊脂糖凝膠電泳驗證擴增結果。
擴增產物經過瓊脂糖凝膠回收純化后,與抽提好的pPIC3.5K質粒同時進行EcoRI與NotI雙酶切,酶切體系:ddH20 23 ILL;TT或pPIC3.5K 20μL;EcoRI IμL;NotI1μL;10xCutsmart 5μL。設置酶切程序:37℃酶切1 h,80℃滅活15 min。酶切產物經由核酸吸附柱回收后,測定濃度。按如下體系加入樣品進行酶連;TT 8.5 μL;pPIC3.5K 8.5 μL;連接酶1μL;10x連接酶緩沖液2μL。設置酶連程序:16℃ 3 h,65℃30 min。酶連完成后全部轉化200μL感受態大腸桿菌中,涂布帶氨芐的LB平板,37℃培養活化12~16 h,挑取轉化子以TF-EeoRI-F與3AOX為引物進行菌落PCR,瓊脂糖凝膠電泳驗證轉化子。挑取能擴增出正確條帶的轉化子于帶氨芐抗性的LB培養液中,放置于搖床37℃,220 rpm培養12~16 h。提取質粒。測定濃度并送去測序。
3、重組酵母菌株的構建與驗證。畢赤酵母感受態制備,質粒的線性化與畢赤酵母的電轉參照文獻2中的方法進行。將長出的畢赤酵母轉化子點到提前準備好的濃度為0.5 g/L的G418抗性平板上,做好標記。30℃培養3~5d后正常生長出的轉化子繼續點到0.75 g/L的G418抗性平板上,逐步提升濃度篩選高拷貝轉化子。用無菌牙簽將高拷貝轉化子挑到5μL無菌水中混勻,放置在PCR儀中95℃處理5min。然后取其中1μL作為模板進行PCR。以5AOX與3AOX為引物,驗證基因是否插入基因組。
4、重組酵母菌株的誘導表達。將高拷貝轉化子及野生型GS115接種至50 mL(500 mL錐形瓶)BMGY培養液中,放置于搖床,30℃下250 rpm培養至0D600=2~6(16~18h),12000 rpm室溫離心5 min收集菌體。用無菌水12000 rpm室溫離心5 min清洗三次去除殘余甘油,最后用100 mL BMMY重懸細胞。于30℃,250 rpm的條件下誘導表達,每24 h取500μL菌液并補加500 μL甲醇。將取得的菌液12000 rpm離心2min,用40 txL無菌水重懸,加入10μL 5×蛋白上樣緩沖液后煮沸10 min,12000 rpm離心10 min,取10μL樣品進行聚丙烯酰胺凝膠電泳,分析目的蛋白的表達水平。
二、結果與分析
(一)目的基因的驗證
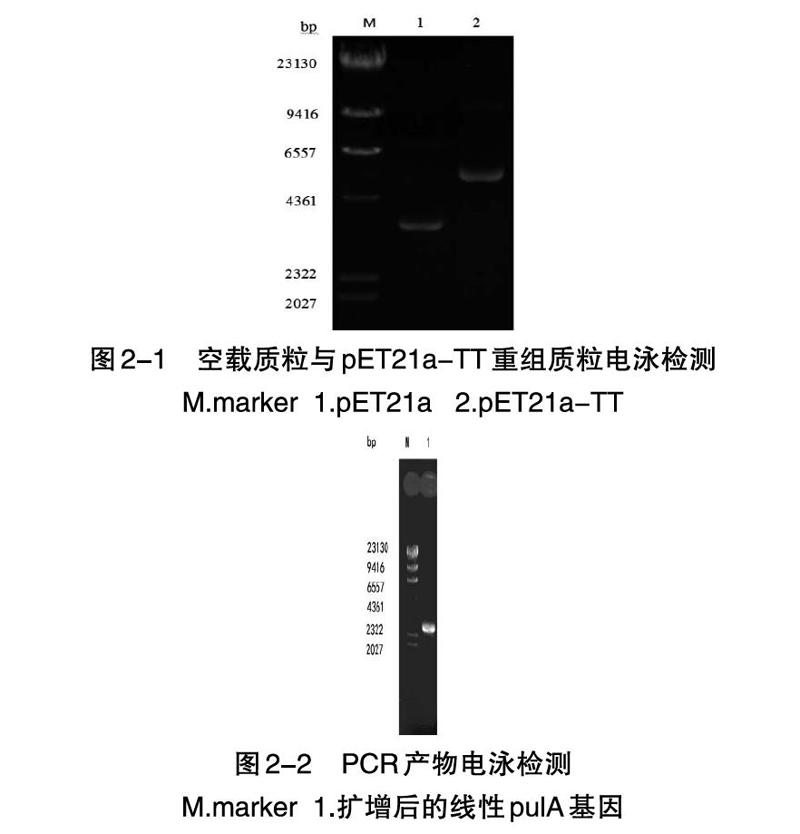
如圖2-1所示,對空載的質粒pET21a和含有普魯蘭酶(pulA)的重組質粒pET21a-TT進行瓊脂糖凝膠電泳檢測,可以看到pET21a-TT因為質量分數較大所以其條帶遷移距離相對于空載質粒更短,且提取的質粒線性條帶位置符合理論值,用軟件DNAman對測序結果的序列進行比對,比對結果發現,測序結果顯示序列是完全正確的。說明含有目的基因的重組質粒經過驗證可以繼續進行實驗。
(二)重組質粒的構建
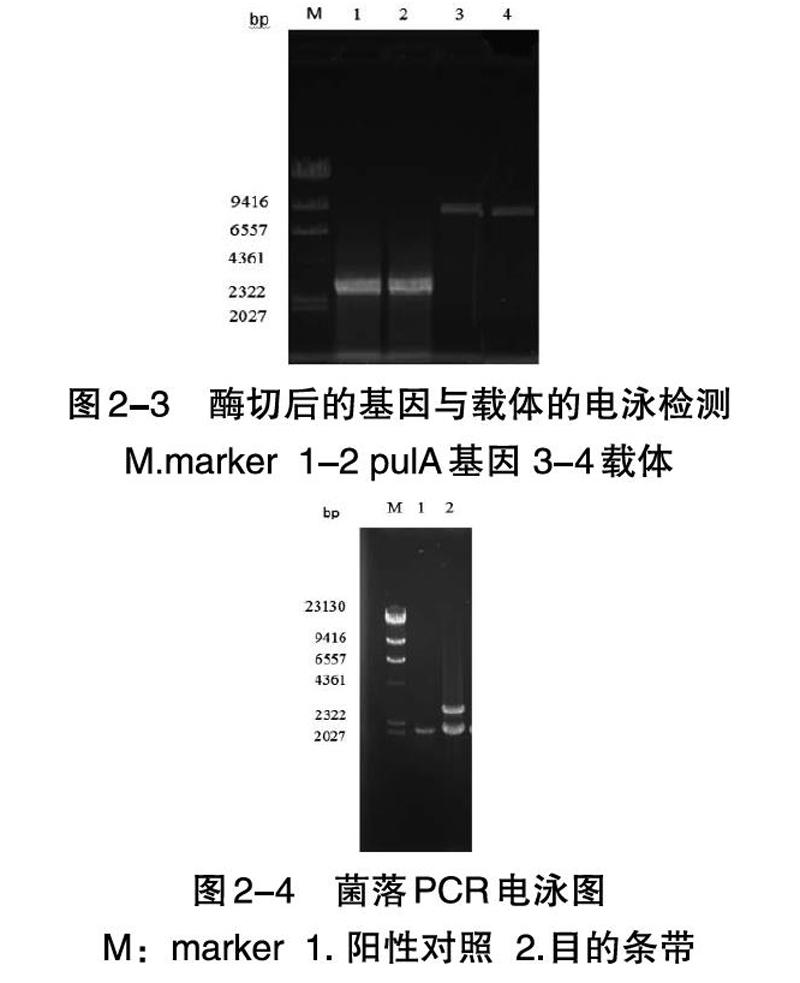
如圖2-2所示,基因的條帶遷移距離相對于Marker條帶的位置符合其序列長度,目的條帶切膠回收后測序結果正確。證明已經成功的擴增出含有雙酶切位點的線性普魯蘭酶基因。如圖2-3所示,基因和載體雙酶切之后進行瓊脂糖凝膠電泳檢測,目的pulA基因大小是2556 bp,載體大小是9004 bp,對照marker條帶的結果,基因條帶位置與預期結果基本相同,證明基因和載體在大小上基本正確,可以進行下一步酶連。
(三)重組酵母菌的構建與驗證
將含有正確重組質粒的大腸桿菌進行大量的質粒抽提,使用SacI線性化后電轉入畢赤酵母,涂布MD平板,30~C放置2~3d長出轉化子。對其中的高拷貝陽性轉化子進行了篩選,挑取能在2 g/L濃度的G418 YPD平板上生長的單菌落進行菌落PCR。菌落PCR電泳結果如圖2-4所示,可以看出擴增出了目的基因條帶,而且在2.1 kb處有明顯的甲醇利用快速型(Mut+)的特異性條帶醇氧化酶1的基因(aoxl)。
(四)誘導表達結果的分析
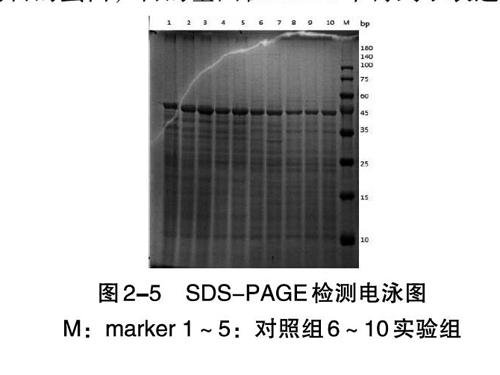
如圖2-5所示,在甲醇誘導過的SDS-PAGE電泳圖中可以觀察到誘導時間為48 h的時候隱約出現一條微弱的條帶,條帶所處的位置對應marker的條帶位置符合目的蛋白分子量,大小是109kDa。說明在該位置出現的可能是本實驗中的目的蛋白,目的基因在GS115中得到了表達。從左至右1~5,6~10依次為每隔24h取得的樣品。
三、結論與討論
本實驗驗證了基因和載體序列的正確性,成功構建了重組質粒,對其進行線性化后轉化畢赤酵母,獲得的轉化子進行鑒定并測序,確認該基因序列正確,成功篩選到陽性轉化子。對畢赤酵母進行普魯蘭酶的誘導表達,采用考馬斯亮藍法對不同時期的菌液蛋白質含量進行SDS-PAGE分析,實驗確定目的蛋白的大小理論值在109 kDa左右。
通過SDS-PAGE分析的結果來看,曾嘗試加大點樣量反復進行實驗,結果仍顯示條帶微弱,蛋白質表達量不高,分析可能是由于Pichia pastoris存在的密碼子偏好性。不同種生物對于密碼子的偏好存在區別。我們的目的基因與Pichia pastoris對于密碼子偏愛性存在著一定的差異。對于Pichia pastoris而言,有25個是其偏愛的密碼子。若本實驗所需要的目的蛋白用到Pichiapastoris中的大量稀有密碼子,可能會對翻譯過程的順利進行產生影響,導致相應的tRNA不足以合成本實驗中的目的蛋白。可嘗試解決的方案為:對密碼子進行優化,將基因進行重新設計,過程中盡量使氨基酸組成與原來的氨基酸組成相同,通過把編碼外源基因的密碼子優化為Piehiapastoris偏愛的密碼子使蛋白表達量增加;選用pPIC9K作為表達載體,嘗試將目的基因進行胞外表達。
