BCR/ABL伴JAK2突變陽性骨髓增殖性腫瘤1例并文獻復習
2020-09-02 06:03:24李俊南朱艷詹茜王利劉林羅小華
解放軍醫學雜志 2020年7期
關鍵詞:基因突變
李俊南,朱艷,詹茜,王利,劉林,羅小華*
1重慶醫科大學附屬第一醫院血液科,重慶 400016;2陸軍軍醫大學西南醫院血液科,重慶 400038
骨髓增殖性腫瘤(myeloproliferative neoplasm,MPN)是一組多能造血干細胞克隆性疾病,2016年WHO修正的新分類中將MPN分為兩類:BCR/ABL基因陽性慢性髓系白血病(chronic myeloid leukemia,CML)和BCR/ABL基因陰性MPN[1]。JAK2基因在BCR/ABL基因陰性MPN發病機制中起著重要作用,尤其在真性紅細胞增多癥(polycythemia vera,PV)、原發性血小板增多癥(essential thrombocythemia,ET)及原發性骨髓纖維化(primary myelofibrosis,PMF)中有較高的致病性[1]。BCR/ABL及JAK2基因常獨立存在于兩種疾病中[2],但臨床發現兩者可同時存在于血液腫瘤患者中,因發生率極低,臨床特點尚未完全明確。現報告1例BCR/ABL伴JAK2突變陽性MPN患者的臨床資料,并對相關文獻進行復習。
1 病例資料
1.1 病史 患者女,66歲,因“體檢發現脾臟增大9年,雙下肢乏力3個月”入院。2007年患者常規體檢行腹部B超發現脾臟增大(具體不詳),未就診。2016年7月患者出現雙下肢乏力,易疲倦,無黑便,2016年10月24日就診于重慶醫科大學附屬第一醫院,并收治入血液內科。起病以來,患者精神、食欲無明顯變化。既往史:否認乙肝、結核等病史,個人史、家族史無特殊。
1.2 入院后體檢 體溫36.3 ℃,脈搏80次/min,呼吸20次/min,血壓110/70 mmHg,體型偏瘦,自主體位,全身皮膚無黃染及淤斑,淺表淋巴結未捫及腫大,腹部膨隆,全腹無壓痛及反跳痛,肝臟肋下未捫及,脾臟肋下5 cm可捫及,質地硬,邊界清楚,表面光滑。
1.3 輔助檢查 血常規:白細胞47.3×109/L,紅細胞2.81×1012/L,血紅蛋白51 g/L,平均紅細胞體積61.2 fl,平均紅細胞血紅蛋白量18.1 pg,平均紅細胞血紅蛋白濃度297 g/L,血小板363×109/L,中性粒細胞百分比78.0%,嗜堿性粒細胞百分比6.0%,網織紅細胞百分比2.1%,網織紅細胞絕對值61.4×109/L。二便常規均正常,肝腎功能、電解質、血脂、血糖正常。
1.4 影像學及其他檢查 腹部B超示:脾臟長徑242 mm,厚約126 mm。骨髓穿刺涂片示:骨髓增生活躍,見大量淚滴狀紅細胞,中性粒細胞堿性磷酸酶(NAP)積分59分。骨髓活檢示:骨髓組織增生活躍,原始幼稚細胞散在可見,纖維組織廣泛增殖。流式免疫分型檢測報告示:異常髓系原始細胞約占全部有核細胞的0.64%,粒細胞比例明顯增高。融合基因檢查示:BCR/ABL(P210)融合基因陽性表達率127.81%,JAK2V617F突變陽性(未定量檢測);染色體:46,XX,t(9;22)(q34;q11)。
1.5 診斷及治療 入院時診斷為慢性髓系白血病合并骨髓纖維化。予以口服伊馬替尼400 mg/次,1次/d,定期復查血常規、腹部B超、骨髓檢測、BCR/ABL及JAK2V617F基因(熒光定量PCR)。6個月后復查骨髓穿刺及活檢較前無改善,基因檢查提示JAK2V617F突變率升高,最終診斷為慢性髓系白血病合并JAK2陽性原發性骨髓纖維化。
2018年8 月3 日(服用伊馬替尼22個月)患者入院查血常規:白細胞4.66×109/L,血紅蛋白72 g/L,血小板352×109/L;腹部B超示:脾臟長徑196 mm;骨髓穿刺涂片示:骨髓增生欠活躍;骨髓活檢示:骨髓增生減低合并纖維組織增生;檢測BCR/ABL融合基因陰性,JAK2V617突變率為73.3%。因此,開始服用JAK2基因抑制劑(盧可替尼)5 mg/次,2次/d,服用1個月左右因肌肉酸軟,自行停藥。但貧血明顯改善,輸血周期延長,血小板明顯下降,脾臟大所致的腹脹癥狀明顯好轉,停用盧可替尼4個月后(服用伊馬替尼27個月)復查JAK2V617F突變率為59.5%。治療過程中患者血常規指標變化如圖1所示,基因突變率、脾臟長徑變化如圖2所示。
2 文獻檢索與復習
在萬方知識服務平臺文獻檢索首頁輸入關鍵詞“慢性髓系白血病/CML”“慢性粒細胞白血病”,在結果搜索關鍵詞中輸入“JAK2”,共顯示52條,篩查到5例簡短中文個案報道。于PubMed-NCBI首頁輸入“chronic myelogenous leukemia”“JAK2”及“BCR/ABL”“JAK2”,可檢索到556篇相關文獻,其中篩查到流行病學研究文獻3篇,個案報道31篇(2篇無法獲取全文)。精讀29篇英文個案文獻,收集有詳細報道的具有BCR/ABL重排及JAK2陽性個案46例,其中男34例,女12例,中位年齡58歲,地域分布:歐美國家27例(德國9例,美國7例,意大利6例,法國3例,英國1例,羅馬尼亞1例),亞洲國家19例(中國7例,韓國6例,日本3例,加拿大2例,馬來西亞1例)。

圖1 治療過程中患者白細胞、血紅蛋白和血小板變化情況Fig.1 Changes of leukocytes, hemoglobin and platelets of the case during treatment process WBC.白細胞;Hb.血紅蛋白;PLT.血小板
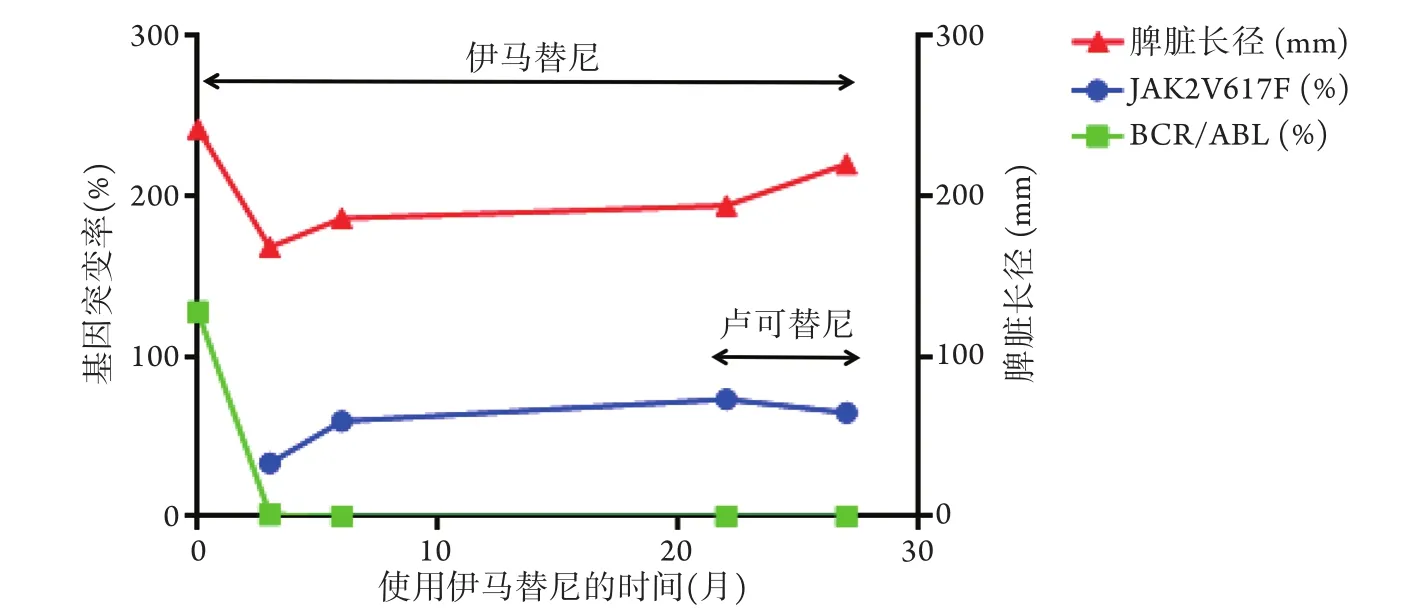
圖2 治療過程中患者BCR/ABL、JAK2基因突變率及脾臟長徑變化情況Fig.2 Mutation rate of BCR/ABL and JAK2 gene and the change of spleen diameter of the case during treatment process
將46例患者按初診診斷及基因檢測結果分為BCR/ABL重排陽性組、JAK2陽性組、JAK2及BCR/ABL雙陽性組。其中13例(28.26%)初診診斷為BCR/ABL重排陽性,治療過程中發現JAK2陽性;14例(30.43%)初診診斷為JAK2陽性,治療隨訪過程中出現BCR/ABL重排;19例(41.30%)初診即發現BCR/ABL重排及JAK2突變陽性。有詳細血常規資料的共33例,方差分析結果顯示,3組患者白細胞、血紅蛋白、血小板數量比較,差異均無統計學意義(白細胞P=0.349,血紅蛋白P=0.247,血小板P=0.944,表1)。
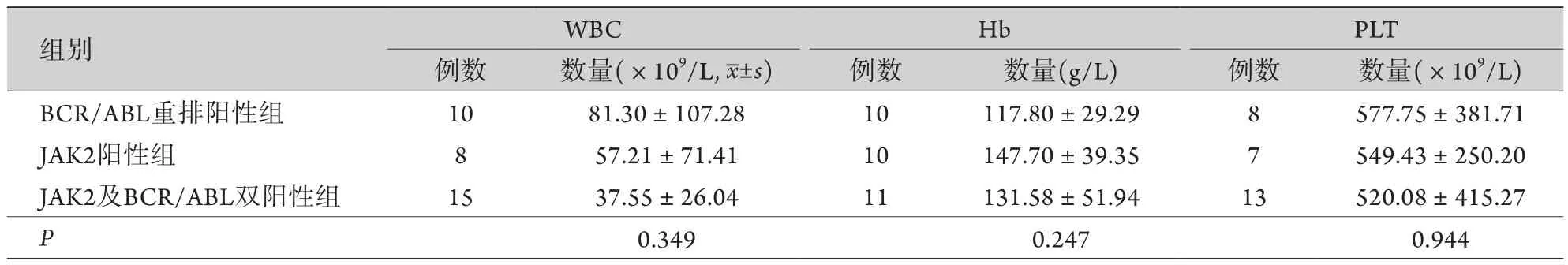
表1 初診時BCR/ABL重排陽性組、JAK2陽性組、JAK2及BCR/ABL雙陽性組血常規指標比較Tab.1 Comparison of blood routine indexes among BCR/ABL(+) rearrangement group, JAK2(+) group and bi-JAK2(+)-BCR/ABL(+) group at initial diagnosis
3 討 論
BCR/ABL伴JAK2基因突變臨床罕見,國外首例于2007年由Hussein等[3]報道。近年來,此類病例引起了重視,多項回顧性研究顯示其發生率為0.2%~4.0%[4-5]。本研究總結既往詳細報道的病例及其臨床特點,以期為臨床診療提供依據。
BCR/ABL及JAK2基因雙陽性患者的臨床分類:①初診BCR/ABL重排陽性,治療過程中JAK2基因由陰性轉為陽性;②初診JAK2基因陽性,治療過程中BCR/ABL重排由陰性轉為陽性;③初診兩個基因均陽性。兩個基因的發生發展可能存在密切聯系,目前有兩種觀點:①單一克隆,JKA2基因為原始突變,BCR/ABL重排為獲得性突變,在發病過程中優勢基因發病;②雙突變克隆雙起源,兩者獨立發生于不同的干祖細胞,突變及發病過程無關聯[6]。有研究發現,JAK2、CALR等基因可以驅動BCR/ABL重排,同時起著調控BCR/ABL基因穩定性和致癌效應的作用[7-8]。Iurlo等[9]對BCR/ABL重排及JAK2基因突變陽性的干細胞進行分選,發現兩個基因多獨立存在于不同的干細胞中。本研究結果顯示,兩個基因出現的先后順序對血常規各項指標無明顯影響,表明兩種基因突變可能是獨立發生的。
既往病例報道顯示,單獨使用酪氨酸激酶抑制劑(TKI)靶向治療時,BCR/ABL基因突變率下降而JAK2基因突變率持續升高,提示TKI對JAK2基因無效,兩個基因可能是相互獨立的。但也有少量報道顯示,在使用伊馬替尼治療過程中,BCR/ABL及JAK2基因突變率同步下降[10];另有4例患者使用達沙替尼治療,其中2例JAK2基因突變率明顯下降[11-12],表明部分TKI對JAK2可能起抑制作用,分析原因為兩種異常基因可能存在于同一克隆中,因此TKI可間接抑制JAK2,但病例數較少,無法確定是否有統計學差異。本病例在加用盧可替尼后輸血次數及脾臟疼痛癥狀均明顯緩解,因此,此類患者中一旦發現有陽性基因,盡早加用相應的靶向藥物,可能對控制疾病進展有重要作用[13-14]。
綜上所述,BCR/ABL伴JAK2基因突變患者臨床罕見,在臨床診治過程中需引起重視,尤其是在對初發疾病進行針對性治療效果欠佳時,需進行積極的篩查。對于JAK2基因是否為BCR/ABL基因的促發基因,目前觀點不一,尚需大樣本的基礎研究證實。BCR/ABL伴JAK2基因突變臨床診療難度大,但目前兩種基因均有相應的靶向藥物,可在一定程度上提高療效,但如何將兩種靶向藥物合理組合、選擇合適的劑量和療程有待進一步臨床研究。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
