復合摻雜BZT 粉體的水解沉淀合成與介電性能研究
2020-09-15 11:46:38劉高斌
陶瓷學報 2020年4期
關鍵詞:結構
袁 琦,劉高斌,韓 露,王 森
(遼寧科技大學 材料與冶金工程學院,遼寧 鞍山 114051)
0 引 言
片式多層陶瓷電容器(MLCC)除具有電容器“隔直通交”的通性特點外,還具備體積小、比容大、壽命長、可靠性高、適合表面安裝等特點,已被廣泛應用于各種軍用、民用電子整機和電子設備,如計算機、電話、程控交換機、精密測試儀器等[1,2]。含鉛鈣鈦礦材料,包括鈮鎂酸鉛、鈦酸鉛等由于介電常數高,曾被大量用作MLCC 的介質材料[3-7],由于鉛有毒且易揮發,對環境污染大,目前,該類材料已經逐漸被鈦酸鋇基陶瓷材料取代。鋯鈦酸鋇[Ba(ZrxTi1-x)O3,BZT]就是部分Ti4+被Zr4+取代后形成的鈦酸鋇基陶瓷固溶體材料。鋯鈦酸鋇材料同鈦酸鋇類似,同樣具有鈣鈦礦結構,在此結構中Ti4+或Zr4+居于Ti-O 八面體中央,Ba2+則處于氧八面體圍成的空隙中。BZT材料介電常數高且不含鉛,介電常數在居里峰附近無突變,使其在大容量電容器介質材料應用方面極具潛力[8-14]。但是,BZT 材料難燒結,介電常數隨溫度變化大,還原氣氛燒結容易變為半導體[15-17]。因此,需要向其中摻雜引入陽離子來進行改性。工業生產中常采用固相摻雜引入,該方法容易造成物料團聚和混料不均,進而影響后續陶瓷介質材料的介電性能;理想的摻雜引入方式為濕化學方法,已見報道的包括溶膠-凝膠法、水熱合成法、草酸鹽共沉淀等。但是,這些方法都有各自的缺點,如溶膠-凝膠法與水熱合成法的前驅體材料成本高,為獲得單一鈣鈦礦結構還需要進一步高溫熱處理;草酸鹽共沉淀法雖然工藝簡單,但是產物中容易夾雜難去除的BaCO3相,而且形成的產物易團聚,燒結過程中易形成二次結晶;而傳統水熱法設備復雜、成本高[3]。這些缺點都限制了傳統濕化學方法在合成理想鋯鈦酸鋇摻雜粉體材料方面的應用。
本文采用水解沉淀合成的方法,制備超細且分散均勻的摻雜鋯鈦酸鋇材料,研究了還原燒成條件下,Nb5+/Ca2+摻雜BZT 陶瓷材料結構和介電性能的變化,討論了摻雜組分對材料結構和性能的影響。
1 實 驗
1.1 粉體合成與陶瓷樣品制備
本實驗所用試劑包括:四氯化鈦(TiCl4),國藥集團化學試劑有限公司;氯氧化鋯(ZrOCl2·8H2O),淄博環拓化工有限公司;氯化鋇(BaCl2·2H2O),國藥集團化學試劑有限公司;氯化鈣(CaCl2·2H2O),國藥集團化學試劑有限公司;五氯化鈮(NbCl5),國藥集團化學試劑有限公司;蒸餾水;自制NaOH 溶液。鋯鈦酸鋇按式BaTi0.9Zr0.1O3合成,Ca2+(由CaCl2·2H2O 引入)在樣品中引入量按樣品3 mol%計,Nb5+(由NbCl5引入)引入量分別按0 mol%、0.1 mol%、0.2 mol%、0.4 mol%計。
粉體合成:將四氯化鈦緩慢滴入到蒸餾水中,保持水溫低于5 °C,同時保持對溶液的強磁力攪拌。依次將氯化鋇、氯化鈣、氯氧化鋯、五氯化鈮的水溶液加入到四氯化鈦溶液中,并繼續攪拌作為前驅體溶液。將自制NaOH 溶液(8 M、16 M)加熱至75 °C,然后緩慢加入前驅體溶液至NaOH溶液中形成沉淀。迅速用蒸餾水反復洗滌沉淀物數次并過濾,以清除其中的氯離子和鈉離子,然后將得到的沉淀物在真空干燥箱中進行烘干。
陶瓷樣片制備:取適量干燥好的粉體,加入5%的聚乙烯醇(PVA)溶液,在瑪瑙研缽中充分研磨進行造粒。然后將造粒好的粉料進行壓片,制成直徑10 mm、厚度3 mm 的圓片,壓力大小為8 MPa,將制好的樣品放于馬弗爐中在800 °C 預燒,冷卻后在樣片兩面涂覆Ni 電極漿料,在管式爐中以3 °C/min 的升溫速度升至1280 °C,爐內通強還原氣氛(N2︰H2=95︰5),保溫180 min,隨爐冷卻。
1.2 樣品結構與性能表征
利用荷蘭 PANalytical 公司 Xpert 型 X 射線衍 射 儀(管電壓40 kv、管電流100 mA)分析粉體產物和燒后陶瓷樣片的物相,射線源為CuKa 靶射線,掃描范圍為2θ= 10-90 °;利用型號為Cambridge S320 的掃描電子顯微鏡觀察粉體形貌和陶瓷樣片顯微結構;利用阿基米德法測量樣片體積密度;采用AT610 型LCR 儀測試樣品介電性能,并依據電容計算材料介電常數。
2 結果與討論
2.1 合成粉體表征
2.1.1 合成粉體的XRD 分析
圖1 示出了不同NaOH 濃度下,Ca2+在樣品中引入量按樣品3 mol%各粉體產物的XRD 譜。圖1a 和b 為NaOH 濃度在8 M,Nb5+含量分別為0 mol%、0.4 mol%條件下所得粉體的XRD 譜,與BZT 標準衍射譜(PDF#31-0174)對比可知,產物主要為四方鈣鈦礦結構的 BZT 相,還有少量的BaCO3(PDF#37-0755),考慮到X 射線衍射檢測出物相體積比一般大于5%,在NaOH 濃度為8 M 條件下會生成一定數量的雜相。圖1c 和d 為NaOH濃度為16 M,Nb5+含量分別為0 mol%、0.4 mol%條件下所得粉體的XRD 譜,與BZT 相標準衍射譜對比可知,各衍射峰位置和相對強度與四方鈣鈦礦結構的BZT 完全對應,產物中未觀測到其它物相。雖然兩種NaOH 濃度環境下,都生成了BZT相,但是從圖1 中可以看出,粉體產物純度受NaOH 濃度影響較大,在本實驗范圍內摻雜Ca2+和Nb5+對粉體物相組成影響不明顯。
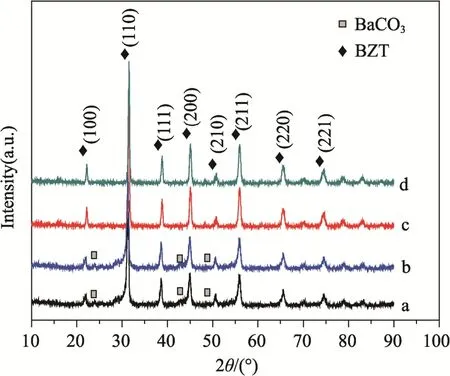
圖1 合成粉體的XRD 譜Fig.1 XRD patterns of the synthesized powders
2.1.2 合成粉體的SEM 分析
圖2(a)、(b)分別為采用8 M NaOH 制得粉體的SEM 圖(Nb5+含量分別為0 mol%、0.4 mol%)。由圖2 可知,所得粉體由兩種物相組成,其中,較大顆粒的相組成為BaCO3,這與XRD 分析結果一致。BZT 相粉體有明顯團聚,分散性差。采用 8 M NaOH 不能制得分散性好且顆粒均勻的粉體。圖 2(c)、(d)分別為采用16 M NaOH制得粉體的SEM 圖(Nb5+含量分別為0 mol%、0.4 mol%),SEM 照片表明,所得粉體為單一物相組成,該物相為BZT 相,形成的晶粒大小均勻、分散性好、無明顯團聚。采用16 M NaOH制得粉體更適合作多層電容器陶瓷介質材料。

圖2 合成粉體的SEM 照片Fig.2 SEM images of the synthesized powders
2.2 陶瓷樣品的結構
鑒于8 M NaOH 制得粉體中存在碳酸鹽且團聚明顯,故僅對16 M NaOH 制得的粉體進行制片,研究其相關結構,Ca2+(由CaCl2·2H2O 引入)在樣品中引入量按樣品3 mol%計,Nb5+(由NbCl5引入)引入量分別按0 mol%、0.1 mol%、0.2 mol%、0.4 mol%計,依次記為BCN0、BCN1、BCN2、BCN4。
不同摻雜濃度的Nb5+粉體所制陶瓷樣品X 射線衍射(XRD)譜如圖3 所示。所有樣品均存在少量BaTiO3-x相,在Ca2+存在的情況下,合成的固溶體中A 位離子過量,在還原氣氛下燒成時,材料內部形成少量氧空位,此時,部分氧重排形成六方結構的BaTiO3-x相,可以消耗部分氧空位,從衍射譜上可以看出,該相與Nb5+摻雜數量無關。與鋯鈦酸鋇PDF 標準卡片對比,發現不同Nb5+摻雜數量樣品的物相一致,衍射峰的位置和相應衍射峰強度都沒有隨Nb5+摻雜數量增加而發生明顯變化,說明Nb5+摻雜不影響陶瓷的主晶相結構,摻雜的Nb5+全部固溶進入晶格。在鋯鈦酸鋇晶體結構中,Ti4+處在體心位置,Ba2+處在晶胞頂角位置,O2-則處在晶胞的六面體面心位置。Nb5+半徑(0.0640 nm)略大于Ti4+半徑(0.0605 nm)。當Nb5+進入Ti4+位替代Ti4+時,根據Goldschmidt 容差因子的計算公式:

式中,t 為容差因子,rA為A 位置陽離子半徑、rO為氧離子半徑,rB為 B 位置陽離子半徑,只要容差因子在0.95 和1.05 間都可以形成穩定的鈣鈦礦結構。Ba2+離子半徑為0.135 nm,O2-為0.140 nm,計算后的容差因子為0.953,Nb5+可以固溶進入Ti4+的位置,形成鈣鈦礦結構,且容差因子由于Nb5+取代Ti4+而減小,因此形成的結構更穩定。
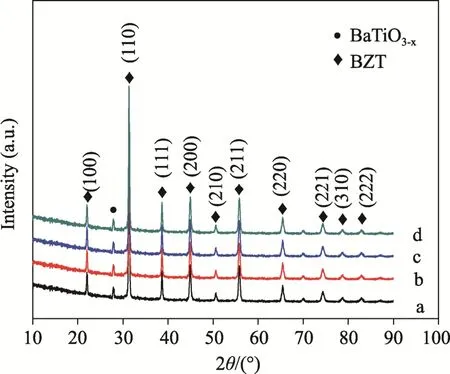
圖3 陶瓷樣品X 射線衍射譜(a) BCN0; (b) BCN1;(c) BCN2; (d) BCN4Fig.3 XRD patterns of the ceramics samples (a) BCN0,(b) BCN1, (c) BCN2 and (d) BCN4
2.3 陶瓷樣片顯微結構分析
為了研究摻雜離子在樣品中對陶瓷材料的結構影響,進行了不同Nb5+含量樣品的掃描電鏡結構分析,如圖4。

圖4 陶瓷樣品的SEM 照片(a) BCN0; (b) BCN1; (c) BCN2; (d) BCN4Fig.4 SEM images of the ceramic samples (a) BCN0, (b) BCN1, (c) BCN2, (d) BCN4
圖4(a)為未摻雜Nb 樣品的掃描電鏡圖。從圖中可以看出,在不含Nb5+的情況下,樣品燒結比較差,有少量氣孔存在,這說明在本實驗條件下鋯鈦酸鋇難燒結,尤其是Ca2+摻雜導致A 位離子過量的情況下。圖4(b)、(c)、(d)分別為Nb5+摻雜數量為0.1 mol%、0.2 mol%、0.4 mol%樣品的掃描電鏡圖,從圖中可以看出,隨著Nb5+摻雜量的上升,樣品平均顆粒尺寸下降,二次結晶雖然沒能完全消除,但數量明顯減少;同時,伴隨著Nb5+摻雜量的上升,材料晶粒內部氣孔和晶間氣孔數量明顯下降,材料的致密性增強。一般來說,由于Nb5+固溶進入鈦酸鋇的晶格中,將取代Ti4+從而導致Ti4+的偏析。該過程產生兩個結果:(1)抑制晶粒進一步長大,由于晶界附近是摻雜離子的聚集區域,會對離子遷移產生釘扎作用,同時阻礙鈦酸鋇相鄰晶粒的晶界遷移,進一步阻礙晶粒的生長。所以隨著少量Nb5+的加入,Nb5+開始在晶界處堆積,由于很難保證少量Nb5+在整個樣品中的均勻分布,因此,當Nb 含量較低時,在高溫長時間保溫后,部分無Nb5+區域出現晶粒二次結晶長大。Nb5+摻雜數量為0.4 mol%的樣品,晶粒二次結晶明顯減少,這主要是由于其廣泛分布于晶界,鈦酸鋇晶粒直接接觸幾率大大下降,晶界遷移困難,因此,大部分晶粒無法通過再結晶長大,Nb5+摻雜抑制了晶粒的生長,同無摻雜或少量摻雜的樣品比,平均晶粒尺寸明顯變小,如圖2(d)所示。(2)由新的液相形成Nb5+取代Ti4+進入晶格位置后,必然會導致少部分Ti4+偏析在晶界附近,這一觀點已經得到其他研究人員的證實[18]。由BaO-TiO2二元相圖可知,BaO 與TiO2摩爾比小于1 后(此種情況出現在Nb 取代Ti 進入晶格后,鈦酸鋇晶界附近),液相形成溫度由1565 °C 立刻降低到1322 °C,考慮到晶界附近可能還有Ca2+等離子存在,實際出現液相的溫度會更低。因此,在燒結溫度為1350 °C 條件下,體系中將出現部分液相。在毛細管力作用下,這些液相會填充到晶粒間隙,可減少材料中的氣孔數量,并伴隨著樣品致密化和體積收縮。
表1 列出了經1350 °C 燒結后摻雜Nb5+數量不同的樣品線收縮和體積密度數值。由表可知,陶瓷樣品的密度隨著Nb5+摻雜數量上升而增大,樣品的收縮率也呈增加趨勢。
E. Brzozowski 等人[19]認為,Nb5+摻雜濃度低時,在材料中存在電子補償機制,摻雜濃度升高時,存在從電子補償向空位補償機制的轉變,此時,Ba 空位的數量快速增加,遠遠超過電子的數量。然而,不論哪種補償機制,隨著Nb5+摻雜數量的上升,都不會導致材料中形成大量的氧空位,材料中的氧空位應主要來自本征缺陷,與Nb5+摻雜數量關系不大。此外,雖然Nb5+的原子量(92)大于Ti4+的原子量(48),Nb5+替代Ti4+占據B 位, 導致體系分子量變大,由于摻雜數量有限,其對密度貢獻并不顯著,密度上升主要與坯體收縮有關。
因此,燒結過程產生的收縮,以及體積密度伴隨Nb5+摻雜數量上升而增加的現象,主要來源于材料中晶界附近少量液相的形成。
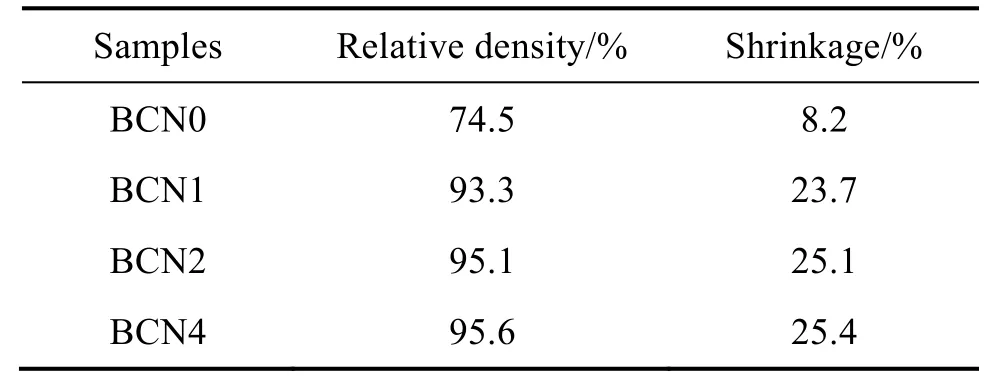
表1 陶瓷樣品相對密度與收縮率Tab.1 Relative density and shrinkage rate of the ceramic samples
2.4 樣品的介電性能分析
純鈦酸鋇為典型的鐵電材料,其居里點即發生鐵電相變的溫度為120 °C,由介溫曲線可知,居里溫度附近的介電常數發生突變,在居里點處介電常數突然升高,當溫度高于居里點后介電常數迅速下降;但是,有摻雜元素時,材料的介溫曲線形狀發生明顯的變化,圖5 為Ca2+/Nb5+摻雜陶瓷樣品的介溫曲線。

圖5 樣品的介電常數-溫度關系曲線(a) BCN0;(b) BCN1; (c) BCN2; (d) BCN4Fig.5 ε-t curves of the samples (a) BCN0, (b) BCN1,(c) BCN2 and (d) BCN4
無 Nb5+摻雜樣品 BCN0 的居里溫度在在75 °C,介電常數在居里溫度附近不再突變,居里峰對應溫區較窄。隨著Nb5+含量增加,居里峰向低溫方向移動,居里峰對應溫區開始變寬,材料表現出明顯的彌散相變特征,隨著Nb5+摻雜數量上升,介溫曲線峰變寬,這與樣品的微觀結構有關。摻雜鈦酸鋇基陶瓷樣品在微觀結構上存在核-殼結構[20],在這種結構中,外來摻雜元素一般在殼結構中的濃度最大,核的部分摻雜元素濃度比較低,即摻雜元素在核-殼結構中呈梯度分布,由外部殼層到內部的核,摻雜元素濃度逐漸下降。在Nb5+含量較少時,介溫曲線的形態主要是由核控制,此時主要是摻雜Ca2+的鋯鈦酸鋇,對應鈣鈦礦的ABO3結構,取代發生在A 位置;Nb5+含量增加后,形成B 位取代,其在殼中的濃度上升,此時,介溫曲線形狀受殼層成分變化和所占比例影響,由于殼對低溫部分材料介電常數的貢獻,從低溫到高溫都有相變發生,介溫曲線變得更寬。此外,無序型固溶體會顯示出明顯的彌散相變,而有序型的固溶體一般不表現出明顯的彌散相變。對于摻雜鈦酸鋇基陶瓷材料,由于其具有復合鈣鈦礦結構,Ca2+或Nb5+分別進入Ba2+或Ti4+的位置,形成的分布是無序的,這種微區的成分起伏是發生彌散相變的原因之一,彌散相隨Nb5+含量上升而增加。材料的晶粒尺寸越大,介溫曲線峰越窄,晶粒尺寸越小,介溫曲線峰越寬且平坦,由于Nb5+摻雜數量的增加,導致晶粒細化,也是介溫曲線峰變寬的一個因素。
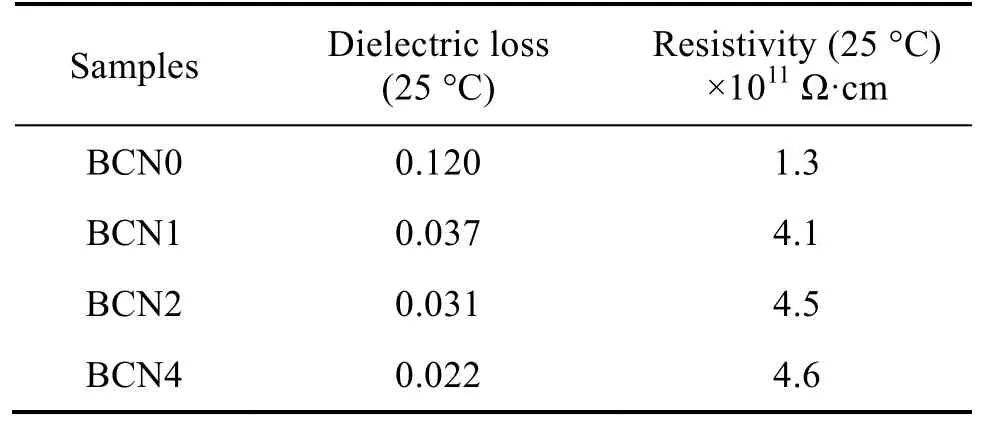
表2 陶瓷樣品的介電損耗和電阻率Tab.2 Dielectric loss and resistance of the ceramic samples
表2 列出了不同Nb5+摻雜量陶瓷樣品的介電損耗和電阻。由表2 可知,添加Nb5+后電阻上升明顯,但是,Nb5+數量增加對電阻值影響不大,其對電阻影響主要是由于提高樣片致密度減少了漏電流,隨著Nb5+含量的上升,介電損耗下降。介電損耗是由于在電場作用下,樣品中少量Ca2+取代Ba2+離子后, 由于Ca2+直徑較Ba2+離子小,導致相鄰的[TiO6]八面體間隙變小;離子半徑比Ti4+大的Nb5+取代Ti4+后,也將會導致少量晶胞體積變化,由此產生晶格的畸變。在這兩種情況的共同作用下,樣品內部會存在少量非鐵電相或鐵電性降低的區域,從而導致極化損耗下降。欒偉玲[21]等人的研究表明,BaTiO3的晶粒粒度也可以影響介電損耗,晶粒粒度越小,損耗也越小,該規律在鋯鈦酸鋇基材料中同樣適用。由于增加Nb5+含量可以有效抑制晶粒長大,因此,隨著Nb5+含量上升,材料的介電損耗逐漸降低。另外,降低氣孔率也可以降低介電損耗。綜上可知,Nb5+摻雜量增加直接導致樣品的收縮率、密度上升,相當于有效降低了材料中的氣孔,能顯著地降低介電損耗。為了降低發熱和漏導,通常要求片式多層陶瓷電容器(MLCC)電介質材料的介電損耗越低越好,因此,向鈦酸鋇中摻雜Nb5+和Ca2+對制備低損耗MLCC 電介質材料有著非常重要的意義。
3 結 論
采用水解沉淀法合成了超細摻雜鋯鈦酸鋇粉體材料,研究了Nb5+、Ca2+離子共摻雜時,NaOH濃度變化和Nb5+含量變化對鋯鈦酸鋇材料的相組成、顯微結構和介電性能的影響。
(1) 通過控制NaOH 濃度,能夠在室溫下合成具有單一鈣鈦礦相的超細摻雜鋯鈦酸鋇粉體材料;
(2) Ca2+的引入促進了六方相BaTiO3-x的生成,該相與樣品中的Nb5+無關;
(3) Nb5+可以固溶進入晶格,取代Ti4+離子,Nb5+半徑略大于Ti4+半徑,當Nb5+進入Ti4+位替代Ti4+時,計算后的容差因子為0.953,容差因子在0.95-1.05 間都可以形成穩定的鈣鈦礦結構,獲得的鋯鈦酸鋇材料相結構不會發生變化;
(4) Nb5+的引入可以促進材料燒結,減少二次結晶,抑制晶粒的長大,另外,Nb5+摻雜可以降低鋯鈦酸鋇材料居里峰并降低介電損耗。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
