Mn-Ce-Pr/Al2O3臭氧催化劑的制備及其性能研究
2020-10-10 01:13:24李重陽程鵬高張建平
功能材料 2020年9期
張 平,項 軍,李重陽,程鵬高,張建平,唐 娜
(1. 天津科技大學 化工與材料學院,天津 300457;2. 天津市鹵水化工與資源生態化利用重點實驗室,天津 300457)
0 引 言
臭氧催化氧化作為一種高級氧化技術(AOPs),其對水體中含有的有機污染物有著十分高效的處理能力,B Legube等[1]調研結果表明,與單獨臭氧氧化相比,臭氧催化氧化與有機物反應速率更高、氧化能力更強同時其幾乎可以完全礦化臭氧單獨氧化無法降解的小分子有機酸、醛等有機物。從添加的催化劑相態可以將臭氧催化氧化劃分為均相臭氧催化氧化和非均相臭氧催化氧化,其中非均相臭氧催化氧化在實際應用中由于所使用的金屬負載性催化劑易回收、對水體的二次污染小以及運行操作方便等[2]特性,使得該項技術成為了這幾年的研究及應用熱門。
為了提高臭氧的氧化效率和利用率,近些年國內外大量研究人員嘗試開發新型高效的臭氧催化劑。其中,葉信國團隊[3]研究表明臭氧在過渡金屬氧化物催化劑的作用下可以有效的降解苯,其中MnO2/ZSM-5在320 min內對于苯的去除率以及礦化率分別高達100%和84.7%;黃元興等[4]則利用30%錳負載量的MnOx/SAC催化劑在pH值=3.5的條件下快速將草酸的去除率從10.3 %提高到92.2 %;汪星志等[5]則利用含錳氧化物的陶粒和普通陶粒為填料,研究臭氧在不同填料條件下對苯甲酸去除效果的影響,結果表明O3/催化劑體系具有更好的催化效果;隆佳君等[6]認為鈰類催化劑在非均相臭氧催化氧化過程中對于部分有機物的降解表現出來了卓越的優越性;王群等[7]研究表明氧化鈰催化臭氧化對于在氧化飾上吸附能力較強的有機物(如鄰苯二甲酸等)有很好的去除效果,可以達到接近90%的去除率;張昂亮等[8]認為Pr負載Al2O3催化劑中的活性成分Pr6O11在非均相臭氧催化氧化過程中對于SA和苯酚有著優良的降解效果,TOC的去除效率可達85%以上;Rodrigo J.G. Lopes等[9]結果表明Mn-Ce-O催化劑在n(Mn)∶n(Ce)=7∶3的條件下對酚類有機物表現出來了卓越的催化效果,僅僅需要60 min就可將酚類有機物全降解;Rui C. Martins等[10]則通過臭氧催化氧化的機理說明了含酚廢水在經過Mn-Ce-O催化劑臭氧催化氧化后不僅獲得了更高的礦化程度,而且最終溶液的生物可降解性也更高;陳茂春等[11]在探討新型多元催化劑催化性能時發現:相同實驗條件下金屬的浸漬順序對催化劑的催化效果有一定的影響,即分步浸漬制得的催化劑的催化效果(50.4%)遠遠好于一步浸漬制得的催化劑的催化效果(45.7%);寧軍等[12]的實驗結果表明,催化劑“MnO2-CuO-CeO2/沸石”的加入可以有效的提高臭氧氧化苯胺的效率,當苯胺初始濃度為200 mg·L-1并且反應20 min后,催化氧化過程將苯胺的去除率由臭氧單獨氧化苯胺的75%提高到了89%。
綜上所述,目前人們主要將目光放在二元體系臭氧催化劑的制備研究上,而對多元體系臭氧催化氧化催化劑的制備和表征研究較少。因此本文首次將過渡金屬與鑭系金屬進行摻雜并制備多元體系催化劑,來提高臭氧催化氧化效率。通過探究浸漬過程、焙燒溫度和負載量等制備條件對催化劑性能的影響,制備Mn-Ce-Pr/Al2O3三元體系催化劑,并采用SEM、XRD、BET以及TGA等表征手段評價所制得的催化劑的理化性質。
1 實 驗
1.1 催化劑的制備
1.1.1 實驗試劑

表1 主要實驗藥品及試劑
1.1.2 苯酚模擬水樣的配置
取10.00 g苯酚溶于500 mL去離子水中并定容于1 000 mL容量瓶中待用;配置的Wt=1%苯酚模擬水樣,其CODCr=1 391 mg·L-1,pH=6.87。
1.1.3 Mn-Ce-Pr/Al2O3臭氧催化劑的制備
本研究“一步浸漬法”制備催化劑的過程分為浸漬與焙燒兩個步驟。
制備催化劑的浸漬過程采用初濕浸漬法[13]:配置不同摩爾比例的金屬鹽混合浸漬溶液,稱取一定質量的Al2O3小球并放在剛好能容納Al2O3小球平鋪靜置的培養皿上,再將配置的浸漬液用一次性滴管逐次滴加在Al2O3小球表面直至其吸附飽和并使浸漬液剛剛沒過其表面,最后用保鮮膜將裝有浸漬液及Al2O3小球的培養皿覆蓋嚴密且繃緊,超聲震蕩2.5 h。
催化劑焙燒前先將浸漬后的樣品用去離子水清洗干凈并在120 ℃的鼓風烘箱中干燥12 h,之后在空氣環境中的某溫度下焙燒5 h。冷卻至室溫,制得Mn-Ce-Pr/Al2O3催化劑。
而“分步浸漬法”制備催化劑的過程就是先按照前面敘述的方法“浸漬+焙燒”制備Mn-Ce/Al2O3催化劑,之后再將Mn-Ce/Al2O3催化劑浸漬在一定濃度的硝酸鐠溶液中,再次按照同樣方法“浸漬+焙燒”制得Mn-Ce-Pr/Al2O3催化劑。
1.2 樣品的性能及表征
通過X射線衍射(XRD)分析確定催化劑的晶型,具體實驗條件如下:工作電壓40.0 kV,立方體靶X射線管電流40.0 mA;根據Brunauer-Emmett-Teller(BET)和Barrett-Joyner-Halenda(BJH)計算總表面積(SBET),總孔體積(Vp)和平均孔徑(Da);熱重分析儀用來研究催化劑在焙燒過程中的失重情況;Mn-Ce-Pr/Al2O3催化劑表面形態通過掃描電子顯微鏡表征;催化劑表面羥基采用滴定法測定[8];CODCr通過哈希水質分析儀器(上海)有限公司的DRB 200 CODCr消解儀和DR 1010 CODCr檢測儀進行測定;水樣pH的變化由pH計精確測定。
2 結果與討論
2.1 實驗裝置
空氣泵、臭氧發生器、氣體流量計、封口反應器和裝有KI的廢氣收集瓶組成了苯酚催化臭氧化的實驗設備,如圖1。在室溫下將100 mL配置的苯酚模擬水樣和1.0 g催化劑放入封口反應器中,由臭氧發生器產生的臭氧在封口反應器的底部曝氣,其中臭氧流速為0.5 L·min-1,臭氧量為1.5 g·h-1,反應時間為30 min。每次實驗結束前,用干燥空氣以2 L·min-1的速度通入處理后的苯酚模擬水樣,持續15 min以消除殘余臭氧對于實驗的干擾。

圖1 催化臭氧化實驗設備簡圖Fig 1 Schematic diagram of experimental equipment for catalytic ozonation
2.1.1 熱重分析
根據1.1.3所述的“一步浸漬法”及“分步浸漬法”分別制備金屬負載量為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0的Mn-Ce/Al2O3催化劑前驅體和金屬負載量為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0.75的Mn-Ce-Pr/Al2O3催化劑前驅體,之后在900 ℃、升溫速率為15 ℃·min-1的空氣氣氛條件下對該前驅體進行熱重分析,如圖2。其中制備Mn-Ce-Pr/Al2O3催化劑前驅體時,中間產物Mn-Ce/Al2O3催化劑是在550 ℃的條件下焙燒制成的。
圖2中兩條熱重曲線分別在548.06 和547.68 ℃時達到了拐點,之后隨溫度升高兩條曲線都逐漸趨于平緩,說明制備Mn-Ce-Pr/Al2O3催化劑的焙燒溫度至少要達到550 ℃,只有這樣才能夠使負載的金屬氫氧化物生成一種穩定金屬氧化物。

圖2 TGA曲線Fig 2 TGA curve
2.2 催化劑的表面形態
通過SEM觀察Mn-Ce-Pr/Al2O3催化劑的表面形態,如圖3,可以看到Al2O3載體在經過金屬鹽溶液的浸漬同時在550 ℃條件下焙燒后,不僅在其表面形成了一層致密的金屬氧化層,并還出現了多孔的結構構造。

圖3 Mn-Ce-Pr/Al2O3催化劑SEM圖(Mn-Ce-Pr/Al2O3孔結構(A);Mn-Ce-Pr/Al2O3表面上的致密金屬氧化層(B);Al2O3載體表面(C、D))Fig 3 SEM diagram of Mn-Ce-Pr/Al2O3catalyst:(a) pore structure of Mn-Ce-Pr/Al2O3; (b) a dense metallic oxide layer on the surface of Mn-Ce-Pr/Al2O3; (c, d) surface of Al2O3 carrier
其原因主要是由于:
(1)浸漬所需的金屬鹽溶液(硝酸鈰和硝酸鐠)屬于強酸弱堿鹽溶液,會在一定程度上刻蝕修飾催化劑載體;
(2)在高溫焙燒條件下催化劑表面的氫氧化物會失水形成該元素的氧化物,使得催化劑表面的金屬氧化層出現凹陷并成孔。催化劑表面的多孔結構不僅會加大催化劑的比表面積,使得催化劑與有機物以及臭氧分子的接觸面積增大,同時孔的出現,為催化劑吸附有機物提供了“溫床”,有利于催化劑催化效率的提升[14]。
同時也表明1.1.3中所敘述的催化劑制備方法是可行的。
2.3 浸漬順序對催化劑催化效率的影響
550 ℃的焙燒溫度下,根據1.1.3中所述的“一步浸漬法”及“分步浸漬法”制備金屬負載量同為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0.25的Mn-Ce-Pr/Al2O3催化劑。以這兩種催化劑催化非均相臭氧氧化1%苯酚模擬水樣同時測定CODCr去除效率,如圖4,發現相同實驗條件下“分步浸漬法”制得的催化劑的催化效率要略高于“一步浸漬法”制得的催化劑,這與文獻[11]所報道的結果一致,即金屬的浸漬順序在一定程度上會影響催化劑的催化效率。

圖4 浸漬順序對催化劑催化效率的影響Fig 4 Effect of impregnation sequence on catalytic efficiency of catalyst
2.4 焙燒溫度對催化劑催化效率的影響
在不同焙燒溫度(450 、550 、650 、750 、850 ℃)下,根據1.1.3中所述的“分步浸漬法”制備5種金屬負載量同為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0.75的Mn-Ce-Pr/Al2O3催化劑。以這五種催化劑催化非均相臭氧氧化1%苯酚模擬水樣同時測定CODCr去除效率,發現相同實驗條件下,五種催化劑的催化效率呈現出隨著焙燒溫度的升高先升高,當溫度超過550 ℃時其催化效率又下降的趨勢,如圖5。
為了解釋這種現象,分別從“催化劑表面負載金屬氧化物的晶型”和“催化劑表面結構”這兩個角度對2.4中的5種催化劑做了表征。
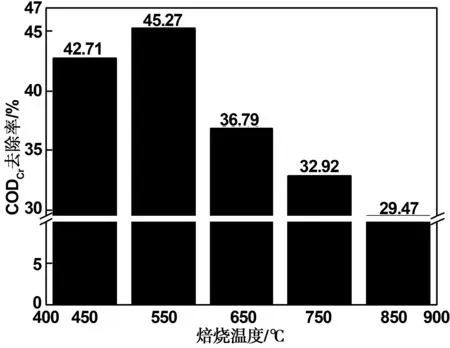
圖5 焙燒溫度對催化劑催化效率的影響Fig 5 Effect of calcination temperature on catalytic efficiency of catalysts
(1)催化劑表面負載金屬氧化物的晶型
對圖2.4中的5種催化劑、n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0的Mn-Ce/Al2O3催化劑以及Al2O3載體進行了XRD表征。在X射線衍射下,5種催化劑均表現出了明顯的金屬氧化物XRD衍射峰,如圖6。同時在圖6中沒有觀察到明顯的金屬氧化物或金屬晶體的XRD衍射峰,這說明各種金屬氧化物良好的分散在了Al2O3表面[11]。與此同時,根據XRD出峰位置可以大致推斷出負載在Al2O3載體上的金屬氧化物與文獻中[7-8、11]所述的有助于提高催化劑催化效率的活性金屬氧化物的組成大致相同,分別為具有活性的Mn2O3、CeO2以及Pr2O3。

圖6 各種催化劑的XRD圖Fig 6 XRD patterns of various catalysts
(2)催化劑表面結構
金屬氫氧化物的熱分解過程一般都是放熱反應,因此適當的提高溫度有利于熱分解反應的進行,但是如果煅燒溫度過高,可能會導致催化劑表面發生板結、比表面積下降以及活性位點缺失的現象,進而降低催化劑的催化效率;而煅燒溫度過低時,金屬氫氧化物又不能發生熱分解反應,形成不了對應的活性氧化物。
圖7是不同焙燒溫度下制備的各種Mn-Ce-Pr/Al2O3催化劑和Al2O3載體的孔徑分布曲線圖,可以直觀的看出:
A、各條孔徑分布曲線都是單峰分布,各種催化劑的孔徑主要集中在2~24 nm之間,最可幾孔徑均約為10 nm左右;
B、隨著焙燒溫度的升高,最可幾孔徑也在慢慢的增加,但是不同焙燒溫度下制備出來的催化劑的孔徑分布曲線峰面積并沒有發生太明顯的變化,這說明這幾種催化劑普遍都具有較大的孔容和較均勻的孔徑分布;
C、根據表2,隨著焙燒溫度的增加,尤其當溫度高于650 ℃時,各種催化劑的平均孔徑(Da)均呈現出顯著增加的趨勢,然而其對應的催化劑比表面積(SBET)卻出現明顯下降,于是得出與文獻[8]中相同的結論:過高的焙燒溫度會使催化劑表面的氧化物發生板結或晶體的塌陷,不利于提升催化劑催化效率。

表2 催化劑的表面積和孔結構

圖7 孔徑分布曲線圖Fig 7 Pore size distribution curves
圖8是不同焙燒溫度下制備的各種Mn-Ce-Pr/Al2O3催化劑的N2吸附-解吸等溫線,各條等溫線均符合Ⅳ型吸附等溫線[15]的特征,因此可以推斷出所有的Mn-Ce-Pr/Al2O3催化劑均為典型的介孔材料。
由于毛細管凝聚現象導致制備的所有催化劑均有明顯的滯后環[16],根據IUPAC的分類:Al2O3載體、Mn-Ce-Pr/Al2O3(450 ℃)和Mn-Ce-Pr/Al2O3(550 ℃)的滯后環屬于典型的H2型,而其余三種樣品的滯后環屬于典型的H1型,這說明全部樣品的孔為大小形狀均勻(H1型)或者大小形狀不均勻(H2型)的狹縫型和瓶型[17]。
圖8中Mn-Ce-Pr/Al2O3(450 ℃)和Mn-Ce-Pr/Al2O3(550 ℃)的吸附支曲線在相對壓力(p/p0)為0.6~0.9處的吸附量均迅速增大,這表明這兩種催化劑具有較大的平均孔徑并且孔徑分布相對集中,都是以2~50 nm的介孔為主,這與孔徑分布(圖7)的結果相一致。
通過這幾種催化劑N2吸附-解吸等溫線的滯后環判斷,在相對壓力p/p0接近1時這幾種催化劑的吸附脫附曲線才可以達到平衡。通常來說,滯后環越大,孔徑和孔容就越大[18],通過圖8可以直觀的看出其余催化劑的滯后環(除Al2O3載體)相較于Mn-Ce-Pr/Al2O3(450 ℃)和Mn-Ce-Pr/Al2O3(550 ℃)催化劑明顯偏小,然而表2表明,Mn-Ce-Pr/Al2O3(650 ℃)、Mn-Ce-Pr/Al2O3(750 ℃)、Mn-Ce-Pr/Al2O3(850 ℃)的孔徑和孔容在增大,這進一步說明過高的焙燒溫度會使負載在載體上的晶體發生板結或塌陷,進而增大了催化劑的孔徑和孔容,同時影響其催化效率。
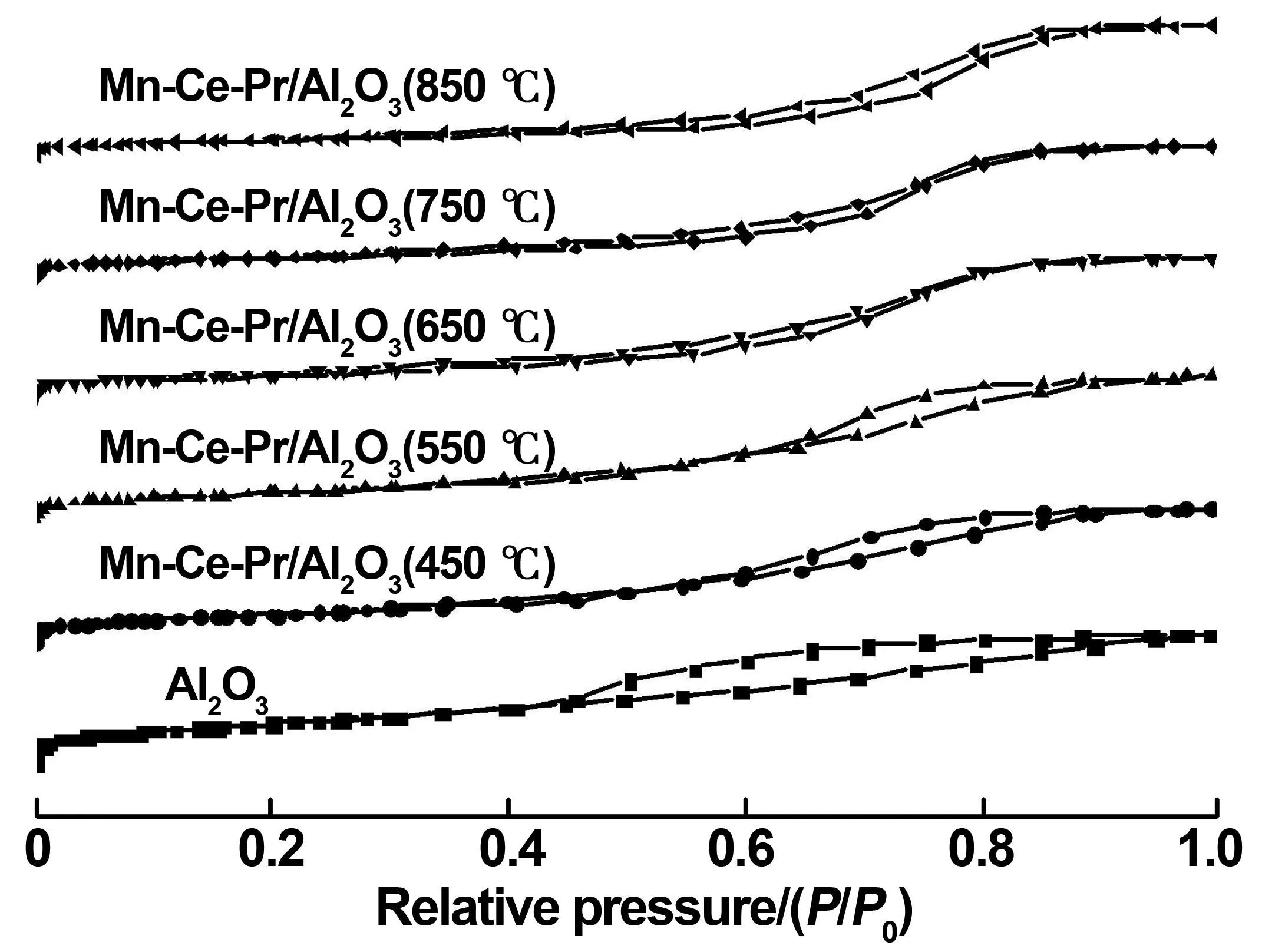
圖8 N2吸附-解吸等溫線曲線Fig 8 N2 adsorption-desorption isotherm curve
2.5 金屬添加量對催化劑催化效率的影響
2.5.1 Ce的添加量對催化劑催化效率的影響
在550 ℃的焙燒溫度下,根據1.1.3中所述的“分步浸漬法”制備金屬負載量為n(Mn)∶n(Ce)∶n(Pr)=(9∶0∶0、9∶1∶0、7∶3∶0、5∶5∶0)的Mn-Ce-Pr/Al2O3催化劑,以這四種催化劑催化非均相臭氧氧化1%苯酚模擬水樣同時測定CODCr去除效率,發現相同實驗條件下,各種催化劑的催化效率隨著Ce添加量的增多呈現出先升高再下降的趨勢,如圖9。
王群[7]利用氧化鈰作為催化劑臭氧催化氧化降解苯類有機物時發現:引入自由基猝滅劑(叔丁醇)后,氧化鈰催化臭氧氧化苯類有機物的速率并沒有降低,而其他非鈰類催化劑催化臭氧氧化苯類有機物的降解速率卻受到了明顯抑制,因此推斷氧化鈰催化臭氧氧化苯類有機物的過程中并沒有自由羥基的產生,由于氧化鈰對氧有很強的吸附能力以及儲氧能力,因此最終得出催化劑表面負載的氧化鈰會增強其對有機物的吸附能力這一結論。同時,王群等[19]又通過一系列的實驗得出結論:相較于任何其他方法,當以CeO2為催化劑分解臭氧時,臭氧分解速率非常快。
綜上所述,Mn-Ce-Pr/Al2O3催化劑在浸漬過程中,Ce(Ⅳ)的加入會增強催化劑的吸附能力從而提升其催化效率,然而過量Ce(Ⅳ)不僅不能提升催化劑的催化效率,相反還會導致反應過程中的臭氧被分解,間接降低了其催化效率。

圖9 Ce的添加量對催化劑催化效果的影響Fig 9 Effect of Ce on catalytic effect of catalyst
2.5.2 Pr的添加量對催化劑催化效率的影響
在550 ℃的焙燒溫度下,根據1.1.3中所述的“分步浸漬法”制備金屬負載量為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶(0、0.25、0.5、0.75、1、1.25、1.5)的7種Mn-Ce-Pr/Al2O3催化劑,以這7種催化劑催化非均相臭氧氧化1%苯酚模擬水樣同時測定CODCr去除效率,發現相同實驗條件下,各種催化劑的催化效率隨著Pr添加量的增多也呈現出了先升高再下降的趨勢,如圖10。

圖10 Pr的添加量對催化劑催化效果的影響Fig 10 Effect of Pr on catalytic effect of catalyst
適當的金屬負載量對于提升催化劑的催化效率至關重要,金屬負載量不足可能會導致催化劑表面活性位點的缺乏進而影響催化劑的催化效率,然而金屬負載量過多則會引起催化劑表面金屬氧化物的簇集,同樣會影響催化劑的催化效率[20]。
根據表面羥基滴定法[8]測得不同Pr負載量的Mn-Ce-Pr/Al2O3催化劑的表面羥基數,其與催化劑催化效率的關系如表3,不難看出,當Mn和Ce的量一定的情況下,隨著Pr負載量的增加,這七種催化劑的表面羥基數雖然在逐漸增加,但是與其對應的CODCr去除效率卻呈現出先增加后降低的趨勢。這說明過多或過少的金屬負載量都會在一定程度上影響催化劑的催化效率,只有在催化劑表面適當的負載活性金屬才可以提升催化劑的催化效率。

表1 不同Pr負載量的Mn-Ce-Pr/Al2O3催化劑的表面羥基數與催化劑催化效率的關系
3 結 論
本研究以Mn、Ce、Pr的氫氧化物為前驅體,粒徑3~5mm的Al2O3小球為載體,同時跟據“初濕浸漬+焙燒”的方法制備了一種Mn-Ce-Pr/Al2O3三元體系催化劑,探究了不同制備條件制備出的Mn-Ce-Pr/Al2O3催化劑催化非均相臭氧氧化1%苯酚模擬水樣時的催化效率。
1)根據“初濕浸漬+焙燒”的方法制備Mn-Ce-Pr/Al2O3催化劑時,“分步浸漬法”比“一步浸漬法”得到的催化劑的催化效率要高;
2)在550 ℃的焙燒溫度下用“分步浸漬法”制備的金屬負載量為n(Mn)∶n(Ce)∶n(Pr)=9∶1∶0.75的Mn-Ce-Pr/Al2O3催化劑具有很高的催化效率;
3)此條件下制備的Mn-Ce-Pr/Al2O3臭氧催化劑表面金屬負載均勻,其滯后環屬于典型的H2型,同時制備的催化劑屬介孔材料;
4)過高的焙燒溫度會導致催化劑表面金屬氧化物發生板結,而焙燒溫度過低則會導致金屬氧化物燒制不完全,金屬負載量過高會導致催化劑表面負載的氧化物發生簇集,而金屬負載量過低又會發生催化劑表面活性位點缺失的情況,因此合適的焙燒溫度和金屬負載量有利于提升催化劑催化效率。
猜你喜歡
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
商周刊(2017年9期)2017-08-22 02:57:49
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國衛生(2014年11期)2014-11-12 13:11:32
