Segawa綜合征1例臨床分析
2020-10-19 14:06:14李丹丹李紅霞何喜寧
中國婦幼健康研究 2020年10期
李丹丹,李紅霞,何喜寧
(陜西省康復醫院兒童康復科,陜西 西安 710065)
Segawa綜合征又稱“多巴胺反應性肌張力障礙”(dopa-responsive dystonia,DRD),于1976年由Segawa等[1]最先報道,是一種罕見的遺傳性運動障礙性疾病,通過常染色體顯性或隱性方式遺傳;其中有一種隱性遺傳的變異型,后來發現是由于酪氨酸羥化酶TH基因突變所致,酪氨酸羥化酶參與了多巴胺的合成,其編碼基因的突變可導致多巴胺合成障礙,從而使體內多巴胺水平降低,引起運動障礙性疾病;但多巴胺受體及其他代謝酶類未受影響,故該病對多巴胺反應良好[2-3]。目前國內外尚無明確的臨床診斷方法,誤診率極高。該病好發于兒童或青少年,對小劑量多巴胺制劑具有良好的反應性,可長期用藥控制病情發展[4]。陜西省康復醫院于2019年2月收治了1例Segawa綜合征患兒,經過基因檢測確診為TH基因突變致病,現報道如下。
1臨床資料
1.1一般情況
患兒,男,1歲5個月,以“發現四肢活動不利5月余”之代訴于2019年2月14日入院。5個月前患兒無明顯誘因出現精神差,緊張時有四肢肌張力增高、頭部扭轉、四肢雙側不對稱、俯臥位不能抬頭、雙上肢不能上舉、不能擺坐、四肢肌肉無力表現。于外院行顱腦磁共振成像(magnetic resonance imaging,MRI)顯示:腦白質局部髓鞘形成欠佳,全脊髓磁共振(magnetic resonance,MR)、肌電圖、腦電圖、人類皰疹病毒(human herpes virus,HHV)、血清氨基酸等檢查未見異常;考慮“腦白質病待查:急性播散性腦脊髓炎?遺傳性腦白質病?”;給予神經節苷脂營養神經、注射人免疫球蛋白抗炎、甲潑尼龍沖擊治療等,癥狀未見改善,且逐漸加重,不能抬頭、翻身。隨后轉至本院。無特殊既往史、個人史、家族史。
1.2入院查體情況
患兒神志清,精神可,面色蒼白,煩躁,易哭鬧,體重11kg,豎頭不穩,不能翻身、獨坐,扶站時雙下肢不能負重,雙足內翻、尖足,雙手無主動抓握意識,拇指內收,無主動語言,不愿與人交流,不能抱奶瓶喝奶,大小便不能示意;心肺腹未見明顯異常,顱神經檢查未見異常;緊張或受刺激時肌張力增高,呈非對稱性姿勢;四肢肌力2級,肌張力:雙側旋前圓肌、內收肌1級,雙側腘繩肌1+級,雙側小腿三頭肌2級,雙側足背屈角-55°~10°;雙側膝腱、跟腱反射亢進,踝陣攣陽性,雙側巴氏征陽性;非對稱性緊張性頸反射、側彎反射陽性,指鼻、對指、輪替試驗不能配合。Peabody評估顯示:姿勢原始6分,相當1月齡;移動原始分3分,相當1月齡;實物操作原始分0分,相當于1月齡;抓握原始分0分,相當于1月齡;視覺-整合原始分13分,相當于2月齡;粗大運動發育商(gross motor quotient,GMQ)為48,精細運動發育商(fine motor quotient,FMQ)為46,總體運動發育商(total motor quotient,TMQ)為43。
1.3輔助檢查情況
血尿糞常規、肝功、腎功、血糖、心肌酶、電解質、心電圖、胸片、骨盆X線未見明顯異常;顱腦MRI顯示雙側大腦白質多發脫髓鞘樣病灶;基因檢測發現患兒TH基因上2個雜合變異,這兩個變異分別遺傳自父親和母親,構成復合雜合變異,見圖1、圖2、圖3。
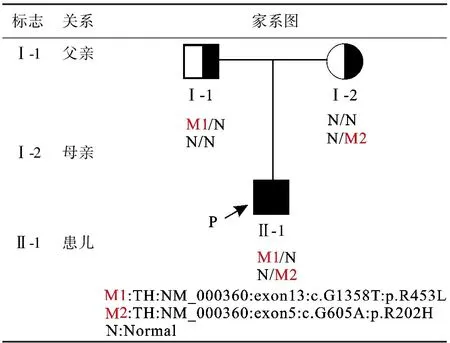
圖1 家系圖
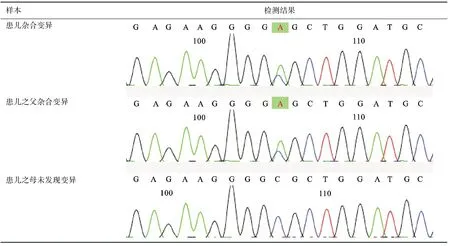
圖2 Sanger測序變異1峰圖(TH:NM_000360:exon13:c.G1358T:p.R453L)
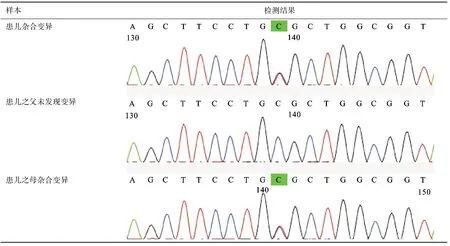
圖3 Sanger測序變異2峰圖(TH:NM_000360:exon5:c.G605A:p.R202H)
1.4治療情況
結合病史、臨床體征和癥狀、輔助檢查結果,該病例確診為“Segawa綜合征”。經團隊評估,制定綜合治療方案,①藥物治療:按左旋多巴3mg·kg-1·d-1給予美多芭(國藥準字號H10930198,左旋多巴200mg和芐絲肼50mg/片)1/6片口服。②綜合康復訓練:a.肢體綜合訓練,按照兒童生長發育順序,采用Bobath療法,通過關鍵點刺激、反射性抑制手法、促通手法等,促通正常姿勢和運動功能發展,訓練量應從小到大;b.多感官游戲治療,緩解患兒焦慮情緒,提高訓練的依從性。③物理因子治療——蠟療:按患兒雙側腘繩肌、小腿三頭肌面積大小,用一次性塑料薄膜包裹加熱石蠟后直接敷于治療部位,每次15分鐘,緩解雙下肢肌張力。④家庭康復延伸指導:醫生、治療師根據患兒訓練情況制定適宜的家庭康復延伸指導,鞏固、延伸康復療效。治療2天后患兒癥狀出現了變化,功能明顯改善,哭鬧焦慮情緒明顯減輕,異常姿勢減少,能豎頭90°維持1分鐘,能翻身、擺坐,雙手可大把抓握。隨后繼續口服美多芭及間斷性康復訓練8個月有余,未出現明顯藥物不良反應。患兒非對稱性姿勢消失,能獨站、獨行,足內翻好轉,足跟可著地,雙手可搭2~3層積木,能用勺子進食,會說疊音或單音詞,會指認五官,可配合穿、脫衣服,可示意大、小便。經Peabody評估顯示:姿勢原始38分,相當18月齡;移動原始分74分,相當14月齡;實物操作原始分5分,相當于13月齡;抓握原始分40分,相當于14月齡;視覺-整合原始分76分,相當于16月齡;GMQ為66,FMQ為73,TMQ為66。
2討論
2.1病因及臨床特點
Segawa綜合征是一種罕見的遺傳病,在原發性肌張力障礙中屬于第五型,其患病率為(0.5~1.0)/100萬[5],女性∶男性為2∶1~4∶1,且女性的外顯率更高。中國Segawa綜合征患者主要以GTPCHI基因突變為主[6]。該病通常在嬰兒期至12歲之間發病,平均發病年齡為6歲,少數在成年期發病;其占兒童和青少年原發性肌張力障礙的5%~10%[7]。但在早期,尤其是嬰幼兒期,由于癥狀不典型易被誤診,如腦癱、早發型帕金森病、發育遲緩及其他類型的肌張力障礙,導致延誤疾病的治療,部分患者確診時已遺留有明顯的后遺癥[8]。
該病遺傳方式有常染色體顯性、隱性及散發,目前已報道致病基因有3個:GCH1、TH和SPR[9]。國外文獻報道顯示,GCH1基因突變所致常染色體顯性遺傳模式平均占65%[10],TH和SPR基因突變所致常染色體隱性遺傳模式占10%~15%;而國內的小樣本家系研究中發現GCH1基因的突變率為70%[11]。本資料的患兒基因突變為TH復合雜合突變,TH基因上有2個雜合變異,變異1為臨床意義未明變異,變異2為致病性變異。
Segawa綜合征臨床表現可大致分為輕、重兩型,輕型表現為1歲以內起病的進展性運動減少——僵硬綜合征及廣泛的肌張力障礙,少數患兒可在1~6歲起病,此類患兒相對于重型而言具有更好的左旋多巴的反應性;重型可表現為圍生期起病的嚴重腦病,晝夜波動的肌張力障礙,通常為清晨癥狀較輕,隨后逐漸加重,常合并有自主神經功能紊亂。患兒長期肢體肌張力障礙可繼發骨骼畸形而致殘,同時兩型都可合并面具臉、震顫、姿勢維持障礙、抽搐、錐體外系相關癥狀及其他帕金森樣臨床特征。本資料的患兒1歲起病,病情進行性加重,突然出現四肢肌肉無力,不能獨站、獨坐,逐漸發展為頭頸四肢主動運動減退,肌張力障礙,足內翻,姿勢不對稱。僅給予其單純的康復訓練,癥狀改善不明顯;結合口服小劑量美多芭后,有明顯療效,符合Segawa綜合征的臨床特點。
2.2治療及預后
Segawa綜合征常易被誤診,需要與肝豆狀核變性、早發型帕金森、脊髓小腦共濟失調等多種疾病鑒別[12],做到早發現、早診斷、早治療。雖然,小劑量多巴胺制劑對Segawa綜合征有戲劇性和持久性反應效果,長期服用左旋多巴無需增加劑量,且不會出現左旋多巴的運動并發癥[13],但迄今為止尚無兒童用藥的標準[14]。有文獻報道,多巴胺制劑的起始劑量通常為1mg·kg-1·d-1,4~5mg·kg-1·d-1時就能最大限度地改善臨床癥狀,如出現惡心、嘔吐、嗜睡、運動障礙、震顫等不良反應則適當減量;早期診治非常重要,對兒童有一定的治愈率,而成人需終身服藥,預后良好[15-16]。
本例患兒,第一天按左旋多巴1mg·kg-1·d-1給予美多芭1/20片(左旋多巴11mg)口服,但家屬未按照醫囑執行;第二天對家屬進行健康宣教,并結合文獻報道[7,16]、患兒的臨床情況及家屬便捷操作需求,試驗性加量,按左旋多巴3mg·kg-1·d-1給予美多芭1/6片(左旋多巴33mg)口服后,效果明顯且持續有效,所以未再繼續加量。在給予患兒綜合康復訓練的同時,指導其家長進行家庭康復訓練延伸,提高患兒的運動、語言、認知及日常生活自理能力,改善其生活質量。
總之,對于早期存在肌張力障礙,臨床擬診Segawa綜合征前,需完善顱腦MRI、血清銅藍蛋白及眼部K-F環等檢查,基因檢測是確診的手段。對于疑似病例,應盡早行相關輔助檢查及基因測序診斷,避免延誤病情。
