基于大數據挖掘與可視化的HPV病毒基因組分析研究
2020-10-27 11:27:38陳翠霞曹宗富李天君于磊喻浴飛蔡瑞琨羅敏娜李乾沈玥陸超高華方馬旭
生殖醫學雜志 2020年10期
陳翠霞,曹宗富,李天君,于磊,喻浴飛,蔡瑞琨,羅敏娜,李乾,沈玥,陸超,高華方,馬旭*
(1.國家衛生健康委科學技術研究所,北京 100081;2.國家人類遺傳資源中心,北京 102206)
宮頸癌是中國女性最常見的惡性腫瘤之一,僅次于乳腺癌[1]。宮頸癌主要病因是高危人乳頭瘤病毒(Human papilloma virus,HPV)的持續性感染[2]。目前,宮頸癌治療手段以毀損性手術為主,缺乏特效無創的阻斷方法,臨床檢測鑒定方法僅覆蓋23種亞型,占現存亞型10%,無法避免假陽性率和漏診率問題,因此有必要對所有HPV進行快速準確的分型檢測[3-6]。最新的疫苗只覆蓋9種亞型的HPV病毒,保護71%的患者[3-6]。隨著高通量測序技術的成熟,病原體的溯源、進化關系、基因組比較、致病危險性預測等研究工作已成為傳染病的防控中重點研究方向[7-10]。有研究工作通過相同亞型HPV病毒的某段基因核苷酸的變異或基因整合熱點位置,來研究病毒進化與疾病的相互關系以及病毒基因整合致病機制[10-13],也有文獻對13種高危亞型的HPV病毒感染后其致病危險性發展的自然史進行監測研究[14]。但仍舊缺乏一種簡單易操作的自動化流程來對HPV基因組大數據進行全面深入的挖掘分析。鑒于此,本文基于比較基因組學研究方法,針對HPV病毒全基因組數據,設計和集成一系列的算法工具包,構建了一種基于大數據挖掘技術的HPV基因組信息可視化分析流程框架,不僅覆蓋迄今為止發現的298種亞型的HPV病毒,且具備對HPV病毒基因組數據的深度比對挖掘能力。
方 法
HPV基因組數據挖掘分析包含3個步驟(圖1):(1)數據預處理:從搜集HPV基因組數據最全面的genBank(genBank[http://www.ncbi.nih.gov/nucleotide/SRA/genBank]數據庫下載所有病毒類基因組數據作為原始數據。采用Perl 5.0語言腳本從原始數據中抽提298個HPV病毒亞型的全基因組數據,及其關鍵核心基因(E6、E7)的核酸和氨基酸序列,采用Perl腳本將關鍵基因按照染色體上的順序連接組裝,建立關鍵核心基因組,全部整理為fasta格式。(2)系統進化分析:采用Dnasp[15]軟件評估全基因組序列的替換飽和度,對于通過評估篩選的HPV全基因組,采用MAFFT[16]軟件進行多序列比對,并采用FastTree[17]軟件的最大似然法完成進化樹的構建,使用Dendroscope 3.0[18]軟件實現全基因組進化樹的展示、編輯和導出,至此完成全基因組進化樹的構建。另外,關鍵核心基因組進化樹的構建方法與全基因組進化樹構建方法相同。(3)氨基酸分類比較分析:基于序列組分和分類比對進行氨基酸表達偏好模式分析。通過Perl腳本統計在HPV關鍵核心基因組的氨基酸序列中,20種氨基酸的占比,然后根據氨基酸的親疏水性、極性、酸堿性分類,匯總為HPV病毒氨基酸分類的占比,用R語言的Scale包對結果進行歸一化校準后得到氨基酸分類矩陣,最后用R語言的Heatmap包繪制氨基酸分類矩陣的熱圖(Heatmap),進而研究基因組分子結構和氨基酸表達偏好模式與毒株生物學特性表型的關系。

圖1 HPV基因組數據挖掘分析框架流程圖
結 果
核酸和氨基酸分子水平結構、氨基酸分類導致物種系統發育進化的不同,進而構成HPV致病危險性和主要感染侵襲部位不同的基礎條件。所以本文從基于分子結構差異的系統進化分析和氨基酸表達偏好模式比較分析兩個方面,來研究他們與亞型分化和致病危險性之間的關系。
1.數據預處理:抽提出上述298種HPV病毒的全基因組和核心關鍵基因(E6、E7)核酸和氨基酸序列,采用Perl腳本將關鍵核心基因按照染色體上的順序連接組裝,建立關鍵核心基因組,全基因組序列與關鍵核心基因組序列均為fasta格式。從這298種HPV中選出致病危險性較為明確的毒株37個和5個侵襲感染類型或致病危險性不太明確的毒株[1-6,10,12-14,19-34],共42個毒株,將其標記感染類型和危險性分類(字母S表示主要侵襲感染皮膚Skin;M表示主要侵襲感染粘膜Mucosa;N表示不明確;HR表示致病性為高危類型High-Risk;LR表示致病性為低危類型Low-Risk)后,與剩余的其他HPV病毒基因組作為參比毒株進入后續分析。統計得知參比的HPV全基因組長度范圍為7 080~8 104 bp。

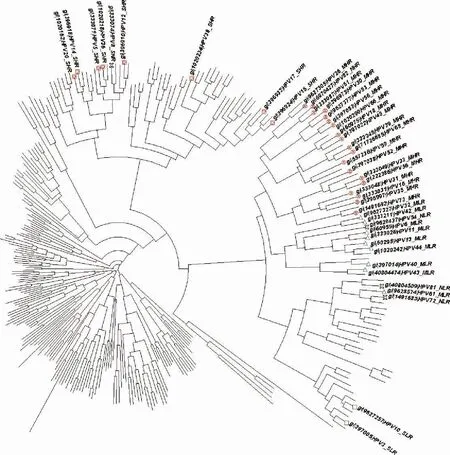
圖2 42個HPV參比毒株在目前現存的298株HPV全基因組進化樹上的分布情況
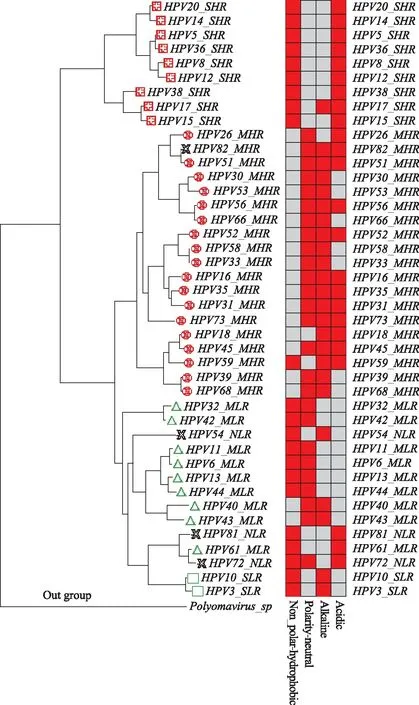
熱圖的紅色塊表示對該類型氨基酸較為偏好,占比較高;灰色塊表示該類型氨基酸占比較少;熱圖第一列是 非極性/疏水性(Non_polar-hydrophobic)類型氨基酸,第二列是極性/中性(Polarity-neutral)類型氨基酸, 第三列是堿性(Alkaline)類型氨基酸,第四列是酸性(Acidic)類型氨基酸圖3 42個參比HPV毒株的關鍵核心基因組進化樹與其氨基酸分類熱圖對應關系圖
3.氨基酸分類比較分析:基于進化樹上毒株的位置,對42個參比毒株的關鍵核心基因組進行氨基酸序列組分和分類比對分析,進而發現氨基酸表達偏好模式與毒株生物學特性表型之間的關系(圖3)。通過Perl腳本分別統計HPV關鍵核心基因組的氨基酸序列中20種氨基酸各自在整條序列中所占百分比,然后將氨基酸分為非極性/疏水性(Non_polar-hydrophobic)、極性/中性(Polarity-neutral)、堿性(Alkaline),酸性(Acidic)四大類,匯總HPV病毒20種氨基酸的百分比為四類氨基酸的占比,然后用R語言的scale包對結果進行歸一化標準化后,得到氨基酸分類矩陣,最后用R語言的heatmap包繪制氨基酸分類矩陣的熱圖(Heatmap),從而可以展示氨基酸表達偏好模式的差異與毒株生物學特性表型的關系。由圖3可見,皮膚高危型(SHR,)毒株比較偏好表達非極性/疏水性和酸性氨基酸產物;黏膜高危型(MHR,)毒株比較偏好表達極性/中性和堿性氨基酸產物,其中毒性較高的HPV16,HPV31,HPV18,HPV35,HPV73,HPV56[14]毒株也同時偏好表達酸性氨基酸;黏膜低危型(MLR,)毒株比較偏好表達非極性/疏水性和極性/中性氨基酸產物,有趣的是HPV40_MLR和HPV43_MLR的氨基酸表達種類模式與MHR的模式很類似,也偏好極性/中性和堿性氨基酸產物,HPV61_MLR和HPV81_NLR的表達模式與SHR類似,也偏好表達非極性/疏水性和酸性氨基酸產物,這也許正是HPV各亞型之間轉化以及低危和高危類型之間轉化的分子水平的物質基礎;皮膚低危型()比較偏好表達非極性/疏水性和堿性氨基酸產物。
討 論
HPV病毒的親緣性或危險性與進化樹上位置密切相關,而進化樹是基于基因組分子組分和結構之間的遺傳距離繪制的。由圖2、圖3可見,HPV病毒根據親緣關系遠近匯聚到不同的簇,聚集位置相近,說明可能是由同一祖先共同進化而來,致病危險性也相似。如果毒株呈現獨立分支,可以預測該毒株是新亞型。本方法曾成功應用于鏈球菌新菌的分離鑒定工作[36-37],而本文將該方法推廣應用到HPV基因組分析,結果顯示全基因組進化樹(參見圖2)的Clade分布趨勢與關鍵核心基因組進化樹(參見圖3)結果相吻合,一方面表明關鍵核心基因E6/E7主導了HPV物種的進化和分化,從分子進化的角度驗證E6/E7蛋白對宮頸癌的發生發展起決定性的作用。同時證明了本流程框架從細菌基因組分型研究[36-37]推廣應用于病毒分型研究仍然是可行的,可以輔助HPV病毒新亞型鑒定、亞型間親緣性和危險性的預測,為HPV病毒的防控提供依據。例如HPV82_NHR,已知屬于高危亞型,但是侵襲部位不很明確,進化樹上與粘膜高危型聚集到一個clade(參見圖2、圖3左),且其氨基酸偏好表達模式與MHR相同(參見圖3右),因此預測其侵襲感染部位為粘膜類型,即為粘膜高危型(HPV82_MHR);HPV8_SNR,已知其屬于侵襲皮膚類型,進化樹上與皮膚高危型聚集到一個clade(參見圖2、圖3左),且其氨基酸偏好表達模式與SHR相同(參見圖3右),因此可以預測其為皮膚高危型(HPV8_SHR);同樣,HPV54_NLR、HPV72_NLR均與粘膜低危型聚集到一個clade(參見圖2、圖3左),同時兩個亞型毒株的氨基酸偏好表達模式與MLR相似,可以預測他們為粘膜低危型,即分別為HPV54_MLR、HPV72_MLR。因此可以說,圖2、圖3從分子進化和氨基酸分類偏好模式角度驗證了文獻[19-22]結果的正確性,致病危險性類似的毒株,其基因組結構特點也類似,找到了HPV病毒的生物學表型特性的差異與核酸、氨基酸分子水平差異有緊密相關性的證據。
基于不同的氨基酸類型在基因組序列中的表達比率,來研究HPV基因組中氨基酸的表達偏好模式,進而發現氨基酸表達偏好模式與病毒侵襲皮膚的特性之間的關系。由圖3可見,致病危險性相似的毒株,氨基酸表達偏好模式也類似,從而保證了其物種基本的生物學特性。MHR和MLR共同偏好表達極性/中性(Polarity-neutral)類型氨基酸,可能說明極性/中性氨基酸的偏好表達與病毒侵襲粘膜的特性有密切關系,而MHR因為更加偏好表達堿性和酸性氨基酸而成為高危亞型,MLR因為更加偏好表達非極性/疏水性氨基酸產物而成為低危亞型;SLR和SHR共同偏好非極性/疏水性,可能表明非極性/疏水性氨基酸的高表達與病毒侵襲皮膚的特性有密切關系,SHR因為更偏好表達酸性氨基酸而成為高危亞型,SLR因為更偏好表達堿性氨基酸而成為低危亞型。基因組核酸分子結構和基因結構的差異,氨基酸偏好表達模式的不同,均導致了HPV病毒侵襲特性的不同和致病危險性的多樣化。
綜上所述,本研究具有以下特點:(1)目前研究最多的HPV亞型數量是131個,本文囊括目前發現的298種HPV亞型毒株代表株,并整理獲得HPV全基因組序列和病毒所有基因(E1/E2/E4/E5/E6/E7/L1/L2)的序列、起止位點、長度、分離病毒的亞型和名稱等詳細信息。(2)通過全基因組比較和系統進化關系分析,可以根據新發病毒在已知致病危險性毒株的進化樹上的分布情況,實現對未知或新發病毒的亞型、感染侵襲特性及其致病危險性的快速預測,為病毒的預防和控制提供證據支持和技術補充。(3)本文也嘗試探究氨基酸表達偏好模式與致病危險性之間的關系,流程框架能夠迅速發現其氨基酸表達偏好模式,利用該表達模式推斷HPV毒株的侵襲特性和致病性危險性,嘗試將基因組結構差異和氨基酸偏好表達模式與生物表型特性做關聯性研究,為指導臨床用藥和控制感染提供依據。
