農藥制劑產品中苯線磷的高效液相色譜分析方法
2020-11-03 02:10:54許春麗馬杜康趙鵬躍黃啟良
現代農藥 2020年5期
關鍵詞:懸浮劑
許春麗,馬杜康,陳 歌,趙鵬躍,黃啟良
(中國農業科學院植物保護研究所,北京 100193)
農藥是重要的農業投入品,農藥產品質量是保障農產品質量安全的重要基礎。然而,近年來一些農藥生產企業為了追求經濟效益,降低生產成本,提高農藥產品防治效果和逃避農藥登記政策等,在農藥產品中添加非法成分(隱性成分或無登記農藥)。而農民傾向于購買防治效果好的含非法添加成分的農藥,這種行為更是助長了農藥生產企業添加非法成分的不良風氣[1-2]。2020年我國農業農村部在全國范圍內部署啟動了農資打假“春雷”行動,重點查處非法添加、含量不足、侵權假冒等問題,以保障農資產品質量。
自2011年10月31日起,我國農業部、工業信息化部、環境保護部、國家工商行政管理總局、國家質量監督檢驗檢疫總局等五部委聯合發出公告,撤銷(撤回)苯線磷等10種有機磷類農藥的登記證、生產許可證(生產批準文件),停止生產,自2013年10月31日起,停止銷售和使用。然而,苯線磷具有較強的殺線蟲效果,雖然已被停止生產,某些企業仍可能在農藥生產中非法添加苯線磷,以增強藥效。
苯線磷(fenamiphos),化學名O-乙基-O-(3-甲基-4-甲硫基)苯異丙基氨基磷酸酯,別名力滿庫、克線磷、苯胺磷等,是一種高毒內吸性殺線蟲劑,兼有觸殺作用。它通過抑制靶標生物中乙酰膽堿酯酶的活性而產生殺蟲效果。原藥雄性大鼠急性經口LD50值為15.3 mg/kg,急性經皮LD50值為500 mg/kg,急性吸入LC50值為110~175 mg/L。在試驗劑量下,對兔皮膚和眼睛無刺激作用,對魚類毒性中等。推薦劑量下,對蜜蜂和蠶無害,對家禽劇毒。目前,在水體和土壤等多種環境介質中均能檢測到殘留的苯線磷及其轉化產物[3]。
有文獻報道利用高效液相色譜法對苯線磷進行分析,可采用外標法定量[4-5],然而,對于農藥制劑產品中苯線磷非法添加的檢測方法尚無報道。筆者在3種代表性制殺蟲劑劑產品(苯氧威懸浮劑、聯苯菊酯乳油、氟蟲腈水分散粒劑)中添加不高于1%的苯線磷原藥,獲得添加苯線磷的農藥制劑試驗樣品,并建立了在同一液相條件下測定苯線磷在懸浮劑、乳油、水分散粒劑中的分析方法,為農藥制劑產品中苯線磷非法添加的檢測提供依據。
1 材料與方法
1.1 儀器與試劑
Agilent 1260-DAD高效液相色譜儀,美國安捷倫科技有限公司;Agilent Eclipse XDB-C18不銹鋼色譜柱(250 mm×4.6 mm,5 μm)、KQ3200B超聲波清洗器,昆山市超聲儀器有限公司;AL-204分析天平,梅特勒-托利多國際貿易(上海)有限公司;有機過濾器(微膜孔徑0.45 μm)。乙腈(色譜純)、二次蒸餾水、苯線磷原藥(質量分數97.1%),農業農村部農藥檢定所;250 g/L苯氧威懸浮劑,江蘇常隆農化有限公司;100 g/L聯苯菊酯乳油、800 g/kg氟蟲腈水分散粒劑,中農立華生物科技股份有限公司。
1.2 液相色譜操作條件
流動相:流動相梯度比例見表1,經濾膜過濾,并進行脫氣。
柱溫:30℃;流速:1.0 mL/min;檢測波長:250 nm;進樣體積:5 μL;保留時間:苯線磷約9.9 min。典型的苯線磷標準品、苯線磷·苯氧威懸浮劑、苯線磷·聯苯菊酯乳油、苯線磷·氟蟲腈水分散粒劑樣品高效液相色譜圖見圖1。

表1 流動相梯度比例
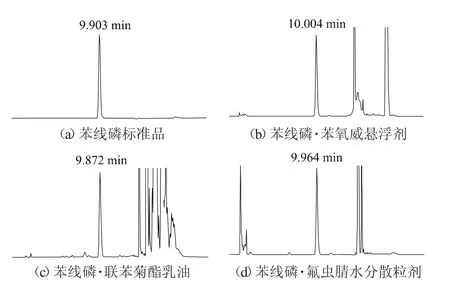
圖1 苯線磷及其3種混劑的高效液相色譜圖
1.3 測定步驟
1.3.1 標準樣品的配制
稱取0.25 g(精確至0.000 01 g)苯線磷標樣于250 mL容量瓶中,加入50 mL乙腈,超聲波振蕩5min,冷卻至室溫,用乙腈稀釋至刻度,搖勻,記為標樣溶液。
1.3.2 試樣溶液的配制
分別稱取24.75 g苯氧威懸浮劑、聯苯菊酯乳油、氟蟲腈水分散粒劑于250 mL容量瓶中,添加0.25 g(精確至0.000 01 g)苯線磷原藥,隨后加入50 mL乙腈,超聲波振蕩5 min,冷卻至室溫,用乙腈稀釋至刻度,搖勻,過濾待測。
1.3.3 測定
待儀器穩定后,在上述操作條件下,連續注入數針標樣溶液,直至相鄰兩針苯線磷峰面積相對變化小于1.2%后,按照標樣溶液、試樣溶液、試樣溶液、標樣溶液的順序進行測定。
1.3.4 計算
將測得的兩針試樣溶液以及試樣前后兩針標樣溶液中苯線磷峰面積分別進行平均。試樣中苯線磷質量分數按式(1)計算。

式中:w為試樣中苯線磷質量分數,%;A2為試樣溶液中苯線磷峰面積的平均值,mAU·s;m1為苯線磷標樣的質量,g;P為標樣中苯線磷質量分數,%;A1為標樣溶液中苯線磷峰面積的平均值,mAU·s;m2為試樣的質量,g。
2 結果與討論
2.1 流動相的選擇
由于懸浮劑、乳油、水分散粒劑干擾較大,為使有效成分得到良好的分離效果,并且分析時間相對合適,峰形尖銳,保留時間適中,根據苯線磷的結構和性質,對不同配比的乙腈和水、甲醇和水2種流動相進行篩選。結果表明,流動相為乙腈+水(體積比45∶55),流速為1.0 mL/min,苯線磷保留時間為9.9 min,能夠獲得良好的分離效果。在苯線磷出峰后仍有很多雜質,為進一步洗脫農藥制劑中的其他物質,在12~17 min將流動相調整為乙腈+水(體積比85∶15)可加快洗脫速度,縮減分析時間。
2.2 檢測波長的選擇
利用紫外-可見檢測器對苯線磷溶液在210~400nm范圍內進行掃描。苯線磷的紫外光譜圖見圖2。從圖2中見,苯線磷的最大吸收波長約250 nm,在該波長處靈敏度較高,各種雜質不影響苯線磷的測定,能夠滿足分析的要求,故將檢測波長確定為250 nm。
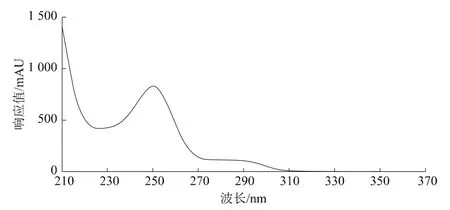
圖2 苯線磷紫外吸收譜圖
2.3 分析方法的特異性
本試驗采用HPLC-DAD峰純度分析法來鑒別苯線磷。苯線磷標樣、苯線磷·苯氧威懸浮劑、苯線磷·聯苯菊酯乳油、苯線磷·氟蟲腈水分散粒劑中的苯線磷HPLC-DAD峰純度均大于990,有效成分處無其他物質干擾,符合定量分析要求。
2.4 分析方法的線性關系
按1.3.1標準樣品的配制方法配制標樣溶液,用乙腈以1∶1的比例梯度稀釋4次,獲得5個濃度的有效成分線性相關溶液。在上述操作條件下,待儀器穩定后,按照分別進同樣體積的標樣,進行測定,取兩次測定的平均結果。以苯線磷質量濃度為橫坐標,峰面積為縱坐標繪制標準曲線(圖3)。從圖3可見,當苯線磷質量濃度為49.34~789.41 mg/L(進樣體積5 μL),其與相應的苯線磷峰面積之間呈現良好的線性關系,計算得回歸方程為y=9.803 3x+35.775,相關系R2=1.000 0,完全可以滿足定量分析要求。

圖3 苯線磷標準曲線
2.5 分析方法的精密度
從同一樣品中稱取5個試樣,在上述色譜條件下進行分析,測得苯線磷·苯氧威懸浮劑、苯線磷·聯苯菊酯乳油、苯線磷·氟蟲腈水分散粒劑的標準偏差分別為0.01%、0.004%和0.01%(表2)。
苯線磷·苯氧威懸浮劑中苯線磷質量分數測定結果平均值為0.93%;變異系數為1.03,小于修改的Horwitz公式的計算值(2(1-0.51ogC)×0.67=2.71,其中C為樣品中有效成分質量分數的平均值)。根據農業部行業標準NY/T 2887—2016《農產品質量分析方法》規范要求[6],有效成分分析方法精密度的測定結果符合要求。
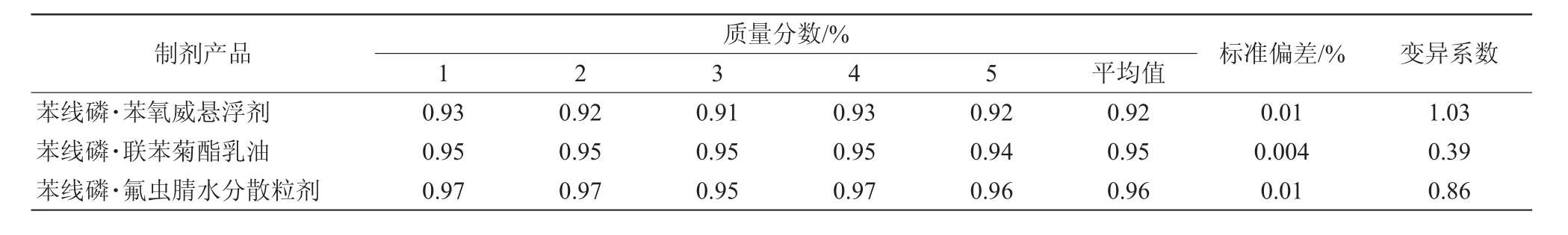
表2 分析方法的精密度試驗結果
苯線磷·聯苯菊酯乳油中苯線磷質量分數測定結果平均值為0.95%;變異系數為0.39,小于修改的Horwitz公式的計算值(2(1-0.51ogC)×0.67=2.70),表明有效成分分析方法精密度的測定結果符合要求。
苯線磷·氟蟲腈水分散粒劑中苯線磷質量分數測定結果平均值為0.96%;變異系數為0.86,小于修改的Horwitz公式的計算值(2(1-0.51ogC)×0.67=2.69),表明有效成分分析方法精密度的測定結果符合要求。
2.6 分析方法的準確度
取按1.3.2試樣溶液的制備方法配制的溶液5 mL于50 mL容量瓶中,加入6 mL按1.3.1配置的苯線磷標樣溶液,用乙腈超聲溶解,冷卻至室溫,再用乙腈定容至刻度,搖勻。苯線磷的回收率按式(2)計算。
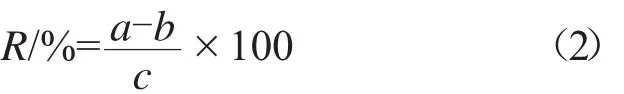
式中:R為回收率,%;a為測定量,mg;b為所稱樣品中待測組分量,mg;c為理論加入量,mg。
經測定,苯線磷·苯氧威懸浮劑、苯線磷·聯苯菊酯乳油和苯線磷·氟蟲腈水分散粒劑的回收率分別為97.61%、99.92%和97.52%,試驗結果見表3,表明有效成分分析方法準確度的測定結果符合要求。
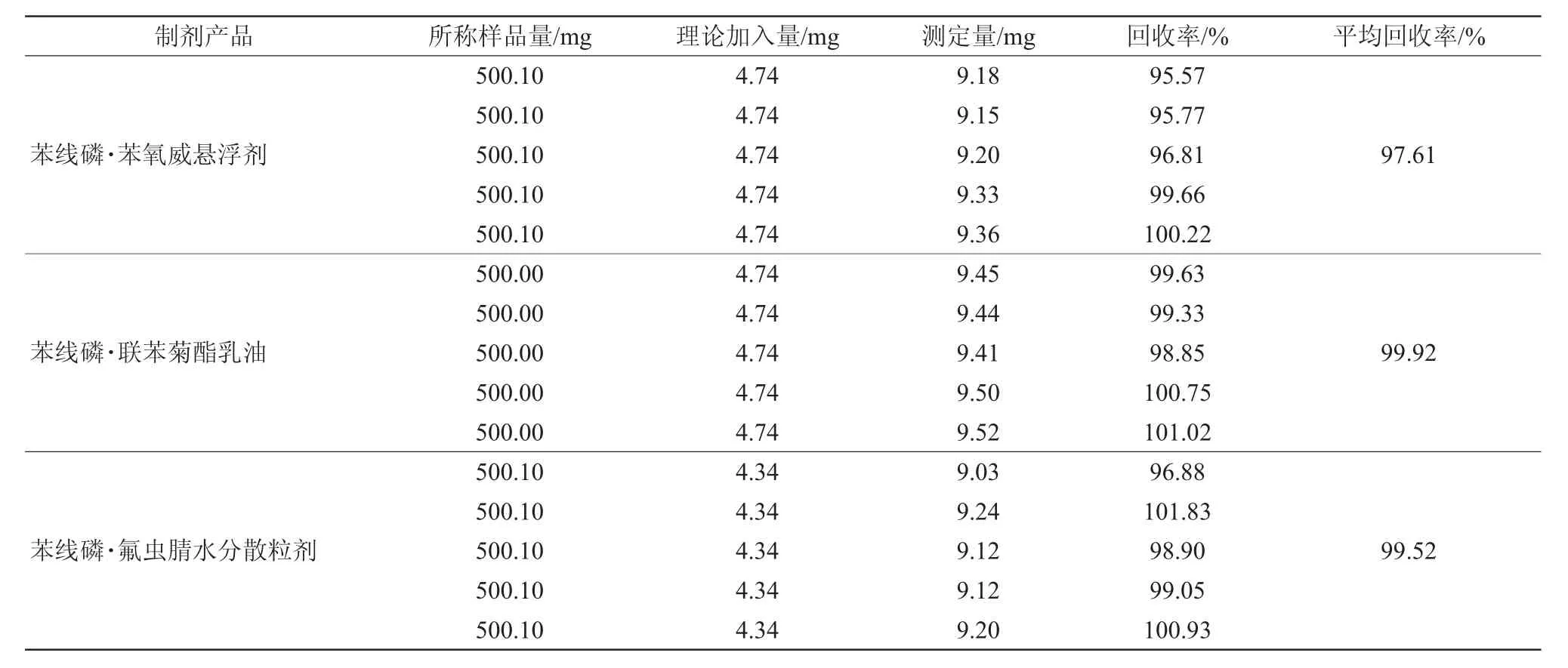
表3 分析方法的準確度試驗結果
3 結 語
利用高效液相色譜對懸浮劑、乳油、水分散粒劑中的苯線磷進行了定性定量分析。結果表明,所采用的方法準確度和精密度較高,線性關系較好,具有簡便、快速、準確及分離效果好等優點,能夠作為農藥制劑產品中苯線磷非法添加的分析測定方法。
猜你喜歡
鉆井液與完井液(2022年4期)2022-10-26 06:39:24
世界農藥(2019年4期)2019-12-30 06:25:10
農藥科學與管理(2019年7期)2019-11-29 07:35:14
農藥科學與管理(2019年9期)2019-11-23 08:41:08
農藥科學與管理(2019年8期)2019-11-23 08:04:44
農藥科學與管理(2019年5期)2019-08-13 00:47:58
農藥科學與管理(2019年10期)2019-04-20 07:13:10
現代園藝(2018年3期)2018-02-10 05:18:13
當代化工研究(2016年7期)2016-03-20 16:21:57
中國果菜(2016年9期)2016-03-01 01:28:41
