
CO2/CH4 干重整轉化催化劑的積碳控制研究
2020-11-16 12:19:08余長春李然家劉慶敬
石油化工 2020年10期
關鍵詞:催化劑
余長春,李然家,王 偉,劉慶敬
(中國石油大學(北京) 新能源與材料學院,北京 102249)
隨著社會經濟的發展及溫室氣體排放帶來的全球氣候變化,基于CO2與CH4的干重整轉化越來越受到研究者的重視。CO2與CH4干重整轉化得到的合成氣通常富含CO,適合羰基合成和氫甲酰化等反應過程,還可以通過干重整與蒸汽重整組合的雙重整生產H2/CO 摩爾比可調的合成氣,用于更多的下游合成,如甲醇。丹麥Topsoe、美國Midrex、日本JOGMNC 等公司都在開發不同應用的干重整工藝技術[1-3]。這些工藝分別使用硫鈍化削弱鎳的活性,以部分蒸汽為氧化劑、貴金屬為活性組分進行干重整反應過程的積碳控制。
由于化學計量的干重整轉化反應在熱力學上是一個難以避開積碳的過程,積碳控制必須從動力學上進行研究[4-11]。干重整轉化的關鍵問題是積碳控制,積碳問題涉及到催化劑和工藝兩個方面。從熱力學上看,干重整轉化的積碳是低溫、高壓、低CO2/CH4摩爾比有利的過程,壓力和CO2/CH4摩爾比都較好控制,通過降低壓力和適當提高CO2/CH4摩爾比可從工藝條件上緩和積碳問題。由于干重整反應是強吸熱反應,需要較高溫度,存在CH4熱分解積碳風險等問題,且反應過程是一個逐漸升溫的過程,不可避免地會遇到對積碳有顯著影響的溫度區間,而干重整轉化反應在中低溫下的積碳問題通常比較突出。因此,干重整轉化積碳控制的關鍵之一是研究中低溫下的積碳控制問題。近年來,受CO2排放與利用問題的影響,干重整轉化的研究也日益成為熱點[12-24]。
本工作采用浸漬法制備了一系列Ni/MgO 催化劑,通過干重整反應對催化劑的性能進行評價,并對評價前后的催化劑進行XRD、SEM、H2-TPR、CO2程序升溫表面反應(CO2-TPSR)等表征,提出了一種比較有效的控制干重整反應積碳的方法。
1 實驗部分
1.1 催化劑的制備
以輕質氧化鎂為載體的主要原料,添加氧化鎂含量為2%(w)的田菁粉,加入一定量的0.1 mol/L 的稀硝酸充分混合,捏合后采用擠條機在高壓下擠條成型,成型的載體經干燥后,采用多段控溫加熱到1 100 ℃,并保持10 h,焙燒得到直徑為3 mm 的成型載體。將成型載體置于1 mol/L 的Ni(NO3)2溶液中充分浸漬1 h,然后抽濾脫除載體中殘余液體,于80 ℃下干燥24 h,并在不同溫度下焙燒,制得催化劑試樣。催化劑中活性組分鎳的負載量主要通過載體吸水率與浸漬次數進行控制。
1.2 催化劑的評價
催化劑評價采用自建微反評價裝置。氣體流量控制采用北京七星華創電子股份有限公司的CS200型質量流量計,評價用氣體為純度99.9%以上的氣體,原料氣和產品氣在改裝成在線雙TCD 的美國惠普公司HP 5890 型氣相色譜進行分析。評價條件:催化劑粒度20 ~40 目,裝填量1.0 g,常壓,原料氣CO2/CH4摩爾比為1.3,甲烷的GHSV=1 000 h-1。反應器為內襯內徑為6 mm 的石英管高溫合金材質反應器。
1.3 催化劑的表征
催化劑程序升溫表征在自建程序升溫/脈沖反應動態表征裝置上進行,采用Pfeiffer 公司QMG 200 型在線四極質譜為檢測器,標準氣體脈沖進行氣體定量。其中,CO2-TPSR 用于催化劑積碳的反應活性表征,O2氣氛程序升溫氧化(O2-TPO)與CO2脈沖結合用于催化劑積碳的定量分析。XRD分析在Bruker 公司D8 Advance 型X 射線粉末衍射儀上進行;SEM 分析在荷蘭FEI 公司 Quanta 200F型場發射掃描電子顯微鏡上進行。
2 結果與討論
2.1 催化劑性能評價與積碳分析
圖1 是Ni 負載量為9.0%(w)的Ni/MgO 催化劑在不同溫度下的反應性能和積碳變化。
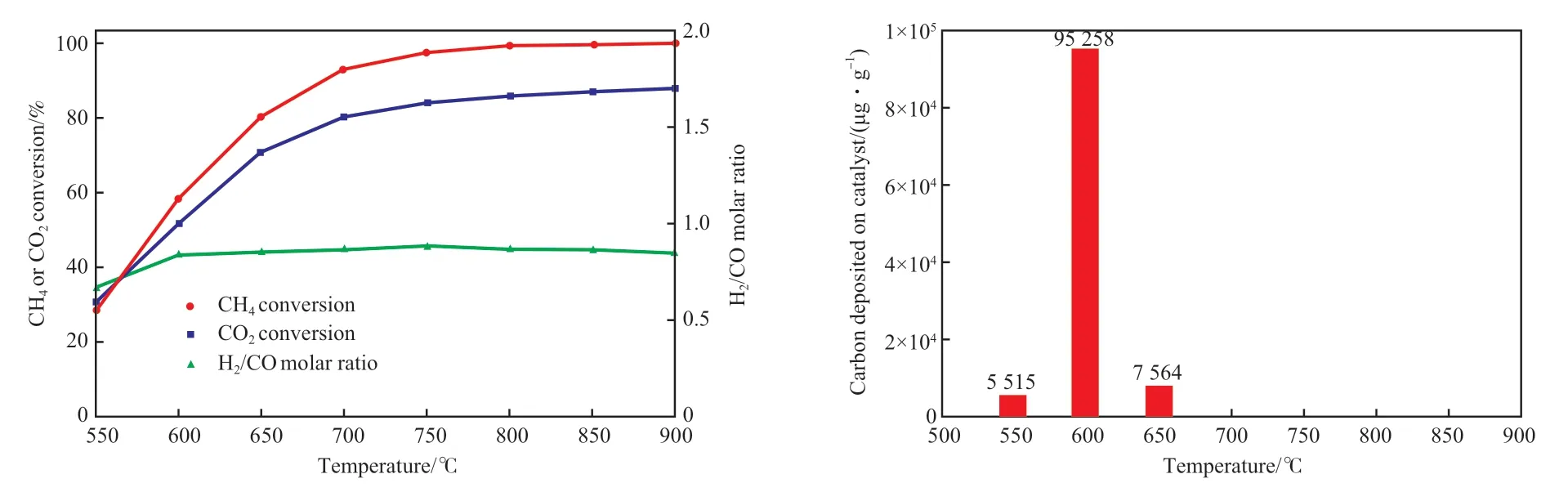
圖1 負載量為9.0%(w)的Ni/MgO 催化劑在不同溫度下的反應性能和積碳變化Fig.1 Conversion and deposited carbon content under different temperatures over a 9.0%(w)Ni/MgO catalyst.Reaction conditions:n(CO2)∶n(CH4)=1.3,GHSV(CH4) = 1 000 h-1,0.1 MPa,20 h.
由圖1 可見,隨溫度的升高,CH4和CO2轉化率逐漸增加,在700 ℃以上時,CH4轉化率明顯高于CO2轉化率,這是因為CO2/CH4摩爾比為1.3時,原料氣中有一部分CO2不發生轉化。550 ℃時,CO2與CH4轉化率相近,主要因為較低溫度下較嚴重的逆水氣變換反應轉化了部分CO2。即使在650 ℃下,H2/CO 摩爾比也一直保持在0.85 ~0.88,比按干重整化學計量反應進行的產品氣的H2/CO摩爾比的理論值1 低,說明較高溫度下存在明顯的逆水氣變換反應,導致部分CO2與H2發生了反應,消耗了部分H2,同時生成了更多的CO。CO 的增加有利于CO 岐化反應,該反應是一個強放熱反應,低溫有利。積碳來源主要有CH4的分解和CO 的岐化。CH4或CO 在活性中心Ni 上的催化解離反應比較容易進行,特別是多個Ni 原子聚集形成的小晶粒上,CH4解離形成石墨結構的碳納米管具有很大的驅動力[25]。同時高溫有利于CH4解離,低溫有利于CO 岐化。600 ℃左右時兩個反應在活性中心Ni 上都比較容易進行,使600 ℃下催化劑上積碳嚴重。
不同Ni 負載量對催化劑干重整轉化反應性能和積碳的影響見表1。由表1 可看出,降低Ni 負載量,即減少表面活性中心數量或密度,能夠明顯降低干重整轉化的積碳量。同時隨Ni 負載量的降低,催化劑的活性也逐漸降低,但降低的幅度較小,說明降低Ni 負載量可以在一定程度上控制干重整轉化過程積碳的形成,但積碳的總量仍較大。這對長周期干重整轉化過程而言,仍存在積碳的問題和催化劑失活的風險,難以用于工業生產過程。
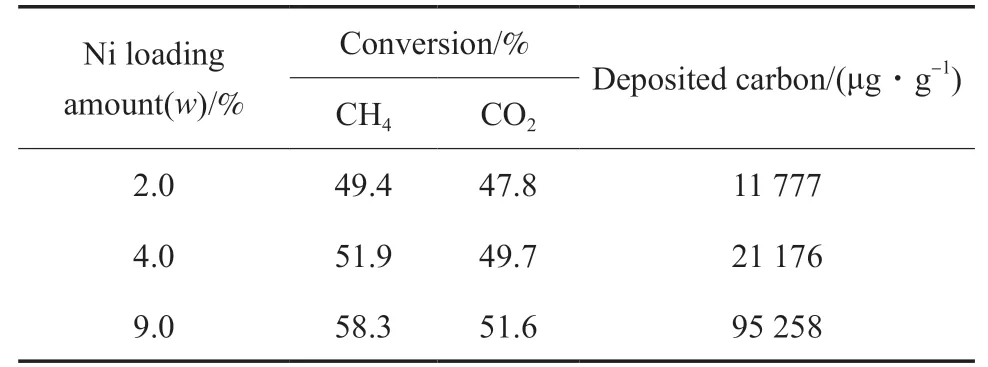
表1 不同Ni 負載量對催化劑干重整轉化反應性能和積碳的影響Table1 The effect of Ni loading amount on the catalytic dry reforming performance and carbon deposition
在Ni 負載量為4.0%(w)時,焙燒溫度對Ni/MgO 催化劑反應性能的影響見表2。從表2 可看出,焙燒溫度對催化劑干重整轉化反應性能和積碳都有很大的影響,400 ℃焙燒的催化劑活性很高,同時積碳量較大。結合表1 結果,說明催化劑的活性中心確實是Ni,對CH4和CO2活化都有效,但對CH4的活化超過CO2。在600 ℃的反應溫度下,CO2的活化比CH4困難,即催化劑低溫活性越高,CH4催化解離的速率越大,且明顯大于CO2活化轉化的速率,這導致催化劑積碳嚴重。400 ℃基本只是負載鎳鹽的充分分解溫度,通常不會與載體產生較強的相互作用,這導致400 ℃焙燒的催化劑上活性中心Ni 能快速活化解離CH4,造成很嚴重的積碳。隨焙燒溫度的升高,催化劑活性明顯降低,特別是950 ℃高溫焙燒的催化劑上沒有檢測到積碳。同時催化劑的CH4和CO2轉化率大幅降低。說明高溫焙燒導致Ni 與載體發生了充分的固相反應,與載體發生了強相互作用。載體主要為MgO,焙燒下Ni 以NiO 形式存在,因Ni2+和Mg2+原子半徑及其氧化物晶體結構相近,可以形成固溶體。因此950 ℃高溫焙燒時,因氧化鎳與氧化鎂固溶體的形成,Ni 與載體氧化鎂形成很強的相互作用,從而削弱了Ni 活化CH4的能力,顯著降低了CH4催化解離反應速率。
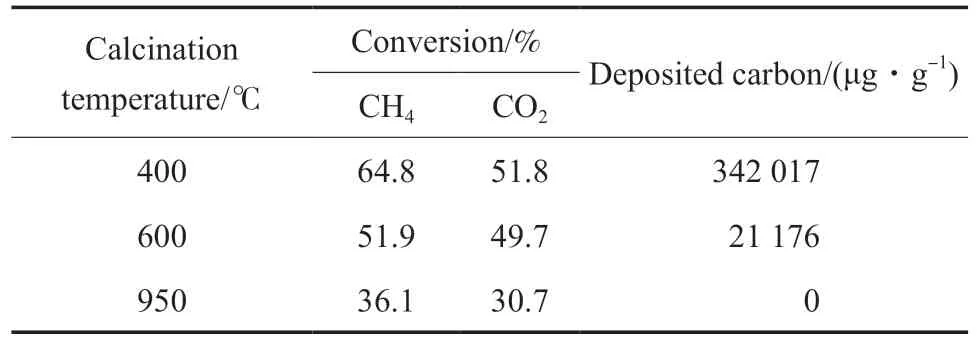
表2 焙燒溫度對Ni 催化劑干重整轉化反應性能和積碳的影響Table 2 The effect of calcination temperature on the dry reforming performance and carbon deposition
2.2 催化劑的表征結果
積碳催化劑的CO2-TPSR 分析結果見圖2。
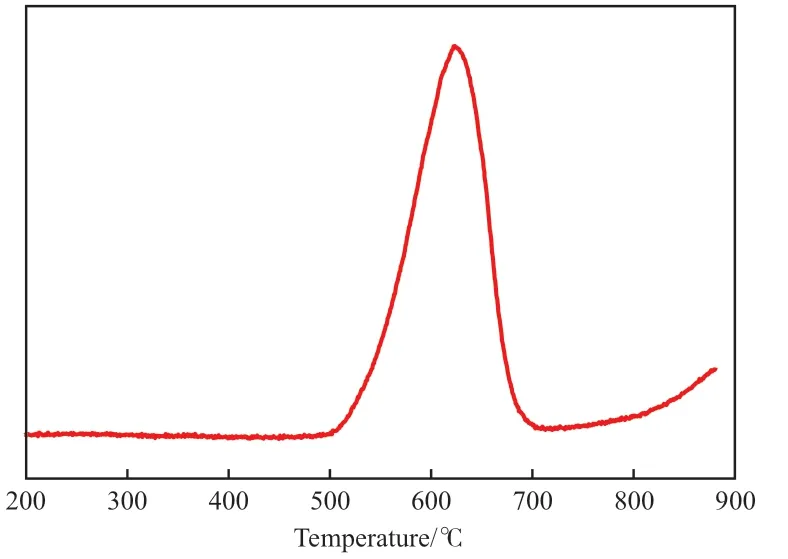
圖2 積碳催化劑上CO2-TPSR 的CO 響應Fig.2 CO response during CO2-TPSR over the carbon deposited catalyst.
由圖2 可見,催化劑上的積碳具有較高的反應活性,在600 ℃時絕大部分積碳可以與CO2反應生成CO,只有很少的積碳需要在800 ℃以上才能與CO2發生反應。結合評價結果可知,該溫度下積碳可能主要來自CH4在活性中心Ni 上的催化解離。
對積碳催化劑和CO2-TPSR 表征后的催化劑進行XRD 和SEM 分析,結果見圖3 和圖4。從圖3 和圖4 可看出,催化劑上的積碳主要是直徑為數十納米的石墨結構的絲狀碳。絲狀碳的衍射峰在進行CO2-TPSR 分析后消失,在CO2-TPSR 分析后的SEM 照片上也沒有觀察到明顯的絲狀碳存在。由此可知:1)直徑為納米尺度的絲狀碳具有較高的反應活性,較容易與CO2發生轉化反應;2)絲狀碳應該主要來自CH4在活性中心Ni 上的催化解離。表面活性中心數量足夠多或密度較大時,CH4的解離速率較快,在CO2/CH4摩爾比為1.3 時,氧化劑CO2難以充分將CH4解離的碳物種轉化為產品氣,導致在催化劑表面產生積碳,說明600 ℃下CO2的活化與轉化較CH4困難,但單獨使用CO2可以充分將這些積碳轉化為CO。這些結果為干重整轉化的積碳控制提供了一個思路,即削弱CH4解離活性或者增強CO2的活化性能。具體方法為:1)強化Ni 與載體的相互作用,削弱Ni 與CH4的相互作用,或者減少表面活性中心Ni 的數量或密度;2)通過提高催化劑表面堿性或者活性金屬電子密度來提高對CO2的活化性能。第一種方法較簡單易行,本研究主要對該方法進行驗證。
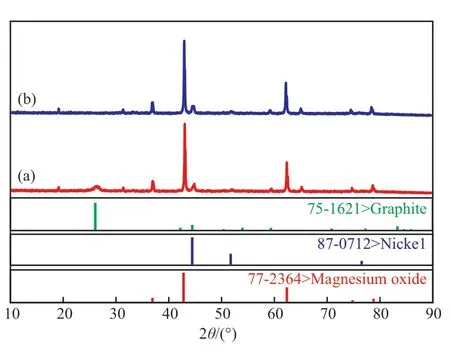
圖3 積碳催化劑在CO2-TPSR 分析前(a)后(b)的XRD 譜圖Fig.3 XRD patterns of the carbon deposited catalyst before(a)and after(b) CO2-TPSR.

圖4 積碳催化劑在CO2-TPSR 分析前(a)后(b)的SEM 照片Fig.4 SEM images of the carbon deposited catalyst before(a) and after(b) CO2-TPSR.
為驗證高溫焙燒是否導致Ni 與載體的強相互作用,對高溫焙燒的催化劑進行了H2-TPR 分析,結果見圖5。從圖5 可見,催化劑在500 ~ 700 ℃區間有兩個弱且寬的還原峰,主要還原溫度在700℃以上,在溫度達1 000 ℃時,H2消耗仍沒有達到峰值,進一步說明高溫焙燒催化劑的大部分Ni 與載體形成了NiO-MgO 固溶體,Ni 與載體存在很強的相互作用。這種Ni 與載體的強相互作用直接削弱了Ni 中心上CH4的活化,導致CH4解離速率大幅下降,即大幅度削弱了CH4的催化解離反應速率,而對CO2與解離生成的碳物種中間物的轉化速率影響較小,結果使得消碳反應速率大于積碳反應速率,從而使得高溫焙燒的催化劑在600 ℃下干重整反應足夠長時間也沒有積碳生成。因此,Ni 與載體發生強相互作用后,不僅削弱了CH4的活化與解離,同時解離形成的表面碳物種與Ni 的作用也顯著削弱,使得這些表面碳物種更容易與CO2反應,轉化為產品氣。此外,大部分Ni 在高溫焙燒下形成NiO-MgO 固溶體,使得活性組分Ni 部分進入體相,得到了進一步的分散,減少了多個近鄰的表面Ni 中心數及其對單個CH4分子活化的幾率,降低了CH4的解離速率。此外,反應氣體CO2是酸性氣體,堿性載體的堿性中心有利于CO2的吸附活化,高溫焙燒形成NiO-MgO 固溶體,Ni 向固相擴散的同時,有利于表面暴露更多的MgO,可以增強CO2的吸附和活化幾率,提高CO2與表面碳物種的反應速率,起到提高消碳反應速率的作用。由此使得干重整轉化過程中CH4催化解離速率小于CO2與解離生成的表面碳物種的反應速率,因此高溫焙燒的催化劑即使在積碳最嚴重的600 ℃下進行反應也沒有積碳生成。
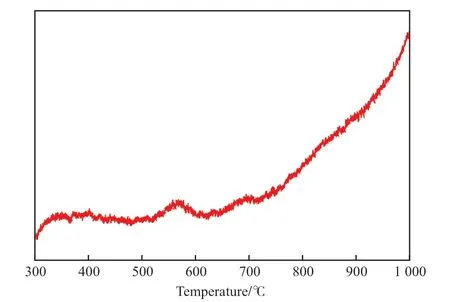
圖5 950 ℃焙燒的催化劑的H2-TPR 譜圖Fig.5 H2-TPR profile of the catalyst calcined at 950 ℃.
3 結論
1)干重整轉化的積碳主要源自在600 ℃的中溫反應條件下CH4催化解離速率大于CO2與CH4解離生成的碳物種反應的速率,積碳反應速率大于消碳反應速率。因此,解決干重整反應積碳的合理方法是通過催化劑的制備來削弱中溫下CH4的解離速率和/或提高CO2的活化與反應性能。
2)通過降低負載量來降低催化劑表面活性中心Ni 的數量或密度,并在400 ~ 600 ℃的相對較低溫度下焙燒制備催化劑,可以在一定程度上削弱CH4解離反應速率,降低催化劑上的積碳量,但并不能有效控制600 ℃下干重整反應過程的積碳。
3)通過提高催化劑焙燒溫度到950 ℃,可以促使大部分活性組分Ni 與載體MgO 形成NiOMgO 固溶體,使得活性中心Ni 與載體MgO 形成強相互作用,提高活性組分Ni 的分散性,表面暴露更多MgO,顯著降低CH4在活性中心Ni 上的解離反應速率,同時有利于CO2的吸附與活化,增強CO2與CH4解離生成的表面碳物種的反應,從而可以有效控制積碳的生成。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
