
載體對固體堿催化劑1,4-丁二醇乙烯化催化性能的影響
2020-11-16 12:19:08武瑞芳呂婷婷王永釗趙永祥
石油化工 2020年10期
關鍵詞:催化劑
武瑞芳,呂婷婷,王永釗,趙永祥
(山西大學 精細化學品教育部工程研究中心,山西 太原 030006)
乙烯基醚聚合物廣泛用于涂料、黏合劑和聚羧酸系減水劑等領域[1-2]。工業上合成乙烯基醚主要采用以液體堿(如KOH、醇鉀等)為催化劑的工藝,該工藝存在副產物多、催化劑分離困難且不可重復使用等諸多問題。創制一種綠色環保的固體堿催化劑,解決液體堿催化劑存在的不足,具有重要的理論意義與實用價值。彭春睿[1]考察了KOH/Al2O3和KOH/AC 等固體堿催化1,4-丁二醇(BDO)乙烯化合成羥丁基乙烯基醚(HBVE)反應,發現盡管KOH/Al2O3催化性能較好,但HBVE 收率也僅為7.1 %。ZrO2具有酸堿兩性和氧化還原性,且熱穩定性高,作為催化劑載體可與活性組分產生協同作用,對許多反應表現出優異的催化性能。張凱等[3]發現Mo/ZrO2催化劑上Mo 物種易于硫化形成MoS2活性位,因而表現出比Mo/Al2O3催化劑更優異的甲烷化催化性能。Li 等[4]將Co/Al2O3和Co/ZrO2催化劑用于費托合成,發現Co/ZrO2因具有適宜的活性組分與載體間相互作用而形成更多Co 活性物種,表現出較好的催化性能。Silvester 等[5]考察了Ni/Al2O3和Ni/ZrO2催化劑的水蒸氣重整催化性能,后者因具有優異的氧化還原性而表現出更高的催化活性。目前,以ZrO2為載體的固體堿催化劑在BDO 乙烯化合成HBVE 反應中鮮見報道。
本工作采用浸漬研磨法制備了K/ZrO2和K/Al2O3固體堿催化劑,通過一系列的表征,考察了不同載體制備的催化劑結構性質與BDO 乙烯化催化性能之間的關系。
1 實驗部分
1.1 主要試劑
ZrOCl2·8H2O:分析純,上海阿拉丁生化科技股份有限公司;Al(NO3)3·9H2O:分析純,天津大茂化學試劑廠;氨水:分析純,上海沃凱生物技術有限公司;硝酸銀:分析純,天津光復科技發展有限公司;KOH:分析純,國藥集團化學試劑有限公司。
1.2 載體及催化劑的制備
稱 取20.921 9 g ZrOCl2·8H2O 溶 于50 mL 去離子水中,在快速攪拌下將10%(φ)的氨水逐滴加到上述溶液中,調節溶液pH=9.5 后繼續攪拌1 h,使它們充分反應,得到Zr(OH)4白色沉淀,室溫下老化12 h,抽濾,同時用蒸餾水洗滌至濾液中檢測不到Cl-為止。抽濾結束后,將沉淀物在120 ℃下干燥8 h,300 ℃下焙燒3 h,制得ZrO2載體。
稱取18.395 9 g Al(NO3)3·9H2O 溶 于50 mL去離子水中,攪拌使其溶解成透明溶液,在劇烈攪拌下將10%(φ)的氨水逐滴加到上述溶液中,當溶液pH=8.5 后老化12 h,抽濾,同時用蒸餾水洗滌至濾液呈中性。抽濾結束后,將沉淀物在120 ℃下干燥8 h,300 ℃下焙燒3 h,制得Al2O3載體。
將4 g KOH 固體和16 g ZrO2載體混合均勻,加入適量的蒸餾水并充分研磨,然后在120 ℃下干燥6 h,再在500 ℃下焙燒4 h 制得KOH 負載量為20%(w)的KOH/ZrO2催化劑,標記為K/ZrO2。按同樣的方法,制得KOH/Al2O3催化劑,標記為K/Al2O3。
1.3 催化劑的表征
采用德國Bruker 公司D8 Advance 型X 射線粉末衍射儀進行XRD 表征,CuKα射線,λ=0.154 06 nm,工作電壓40 kV,工作電流40 mA,掃描范圍2θ=10°~80°,掃描速率6(°)/min;采用美國Micromeritics 公司ASAP-2020 型物理吸附儀進行低溫N2吸附-脫附表征,催化劑預先在150 ℃真空條件下脫氣預處理5 h,然后在液氮浴下進行測定;采用德國Bruker 公司Tensor27 型傅里葉變換紅外光譜儀對催化劑進行FTIR 表征,催化劑與溴化鉀以質量比為1∶100 混合,研磨制片,室溫下進行測定,分辨率4 cm-1;采用德國NETZSCH 公司STA449C 型熱重分析儀進行催化劑的TG 表征,將8 mg 催化劑放入坩堝中,以10 ℃/min 的升溫速率從35 ℃升至800 ℃,載氣為高純N2,流量為60 mL/min;采用ESCALAB 公司250Xi 型光電子能譜儀進行XPS 表征,使用AlKα射線(hν=1 486.6 eV),結合能以碳的C1s的結合能284.8 eV 為基準進行校正;采用Varian 公司710ES 型電感耦合等離子體原子發射光譜(ICP-AES)儀確定反應濾出液中K+的濃度,平行測定3 次,結果取平均值;采用美國Micromeritics 公司Autochem Ⅱ2920 型化學吸附儀進行CO2-TPD 表征,將0.1 g 催化劑(40 ~60 目)置于反應管中,先升溫至300 ℃,N2處理1 h,然后降溫至50 ℃,將N2切換為高純氦氣,流量30 mL/min,同時采用脈沖法多次注入CO2至催化劑吸附飽和,以10 ℃/min 的升溫速率升至700 ℃,TCD 檢測,以未進行CO2吸附的催化劑升溫過程檢測的信號為背景。
1.4 催化性能測試
在裝有溫度計、導氣管與冷凝管的三口燒瓶中加入3 g 催化劑和20 g BDO,啟動磁力攪拌器,攪拌轉速為400 r/min,同時通入N2并開始加熱,N2流量為30 mL/min,待反應溫度升至150 ℃時,將N2切換為乙炔,調節乙炔氣體流量為40 mL/min。反應9 h 后,停止加熱,將乙炔切換為N2進行保護降至室溫。為降低實驗誤差,每個催化劑上催化性能測試平行做3 次,結果取平均值。
1.5 反應產物HBVE 的定量分析
采用Agilent 公司7890B 型氣相色譜儀進行試樣分析,柱型為OV1701,氣化溫度為240 ℃,檢測溫度為240 ℃;柱室溫度為初溫80 ℃,保溫3 min,升溫速率為20 ℃/min,終溫為220 ℃;進樣量0.02 μL;歸一化法定量分析。
2 結果與討論
2.1 催化劑的XRD 表征結果
圖1a 為載體及對應催化劑的XRD 譜圖。由圖1a 可 知,Al2O3在2θ=14.3°,28.5°,37.6°,39.3°,46.2°,67.3°處出現峰形相對寬化的衍射峰,分別歸屬于γ-Al2O3的(111),(220),(311),(222),(400),(440) 晶 面(JCPDS No.10-0425)。與載體Al2O3相比,K/Al2O3催化劑中Al2O3的衍射峰強度有所減弱,表明結晶度降低。這說明催化劑中KOH 并不是簡單地負載在Al2O3載體表面,而是與Al2O3發生了相互作用,Al2O3結構被KOH 破壞,從而導致結晶度有所降低。值得注意的是,K/Al2O3催化劑在30°~33°區間內出現了新的非常彌散的衍射峰,可歸屬于K—O—Al 物種的特征衍射峰[6],說明焙燒過程中,催化劑上部分KOH 與載體Al2O3相互作用形成了KAlO2物種。載體ZrO2僅在2θ=31°處出現了一個微弱的衍射峰,表明ZrO2以無定形態存在。與ZrO2相比,K/ZrO2催化劑在2θ=30.3°,35.5°,50.9°,59.6°,62.2°處出現了明顯的衍射峰,分別歸屬為四方相ZrO2的(011),(110),(020),(013),(211)晶面(JCPDS No.17-0923)[7]。可見,K/ZrO2催化劑經500 ℃焙燒后載體由無定形態轉變為四方相。四方相ZrO2的形成使得KOH 與ZrO2載體的相互作用難以體現。圖1b 給出了經500 ℃焙燒后ZrO2載體(500-ZrO2)的XRD 譜圖。由圖1b 可知,500-ZrO2晶體結構亦呈四方相,但K/ZrO2催化劑中四方相ZrO2的特征衍射峰明顯減弱,同時峰位置有所偏移,原因同樣是由于焙燒過程中KOH 與ZrO2發生相互作用,且部分K+進入ZrO2晶格。K/ZrO2催化劑的XRD 譜圖中未觀察到類似于KAlO2的新物相,這是由于ZrO2固有空位結構小于γ-Al2O3,因此推測只有部分K+嵌入ZrO2載體表面的空位中形成了K—O—Zr 物種[8]。此外,K/ZrO2和K/Al2O3催化劑的XRD 譜圖中均未檢測到KOH 的特征衍射峰,這表明未進入載體晶格的KOH 物種高度分散于載體表面。
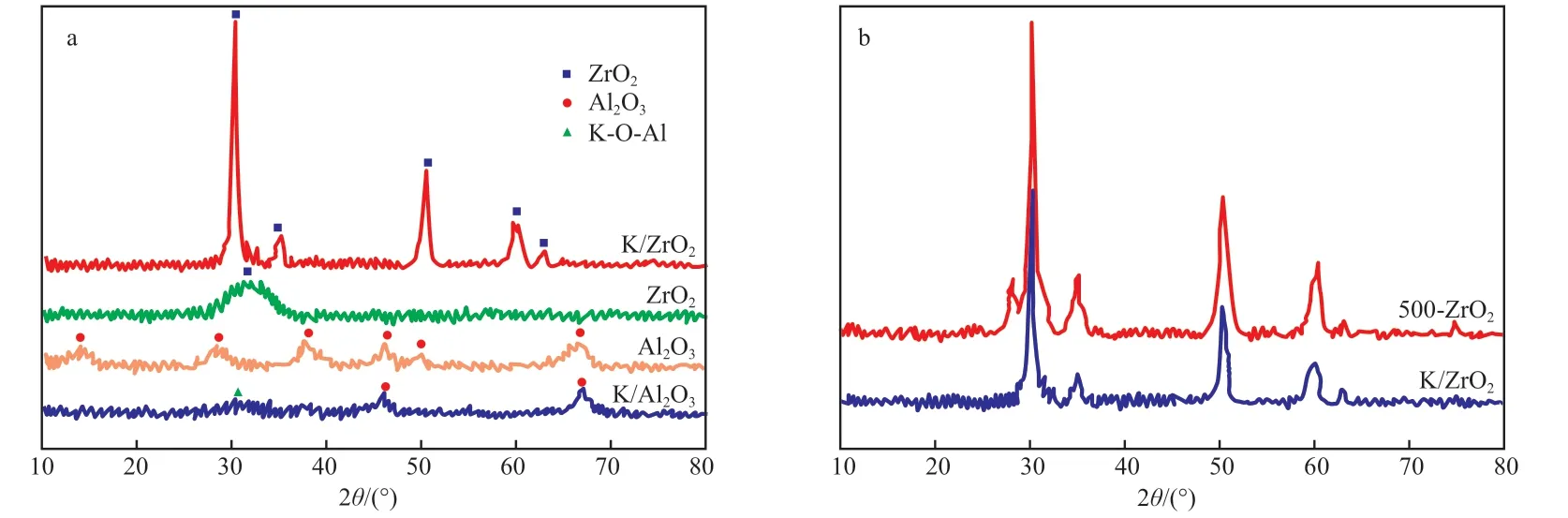
圖1 載體和催化劑的XRD 譜圖Fig.1 XRD patterns of supports and catalysts.
2.2 催化劑的N2 吸附-脫附表征結果
表1 為載體及催化劑的織構性質。由表1 可知,載體的比表面積差別較大,其中ZrO2的比表面積為142 m2/g,而Al2O3的比表面積則高達345 m2/g。與載體相比,在負載KOH 后,催化劑的比表面積均顯著降低。K/Al2O3的比表面積從345 m2/g 急劇下降到54 m2/g,而K/ZrO2的比表面積也僅有8 m2/g。同時,催化劑的孔體積也明顯減小,但平均孔徑卻增大。這主要是由于一方面在催化劑制備過程中,負載的KOH 分布在載體表面以及孔道中,堵塞了載體中原有的部分孔道;另一方面為了更好地形成穩定的堿性位點,催化劑的焙燒溫度相比載體進一步提高,更高的焙燒溫度引起載體不同程度的燒結和部分孔道坍塌;此外,根據XRD 表征結果可知,KOH 的強堿性對載體結構,尤其是Al2O3載體結構也造成了一定的破壞。
2.3 FTIR 表征結果
圖2 為K/Al2O3和K/ZrO2催 化 劑 的FTIR 譜圖。由圖2 可知,K/Al2O3催化劑在3 430 cm-1處附近出現了明顯的紅外吸收峰,這歸因于催化劑表面物理吸附水及自身表面—OH 的伸縮振動[8]。1 630 cm-1處出現的吸收峰是由H—O 鍵彎曲振動所致[9]。1 526 cm-1處的吸收峰歸屬于Al2O3垂直縱向聲子吸收振動[10],1 384 cm-1處的吸收峰歸屬于配位羥基的吸收峰[8]。此外,在757,584 cm-1處出現的紅外吸收峰歸屬于Al—O 鍵的振動[11]。K/ZrO2催化劑在3 430,1 630,1 384 cm-1處也分別出現了紅外吸收峰,同樣是由催化劑表面物理吸附水和表面羥基的伸縮振動、彎曲振動引起的。500 ~1 000 cm-1之間出現的紅外吸收峰歸屬于四方相ZrO2中Zr—O 鍵的伸縮振動[12-13],這與XRD 表征結果相一致。與K/Al2O3相比,K/ZrO2催化劑在3 430 cm-1處附近的吸收峰強度顯著減小,可見K/ZrO2催化劑上的吸附水和表面羥基數量少于K/Al2O3催化劑。
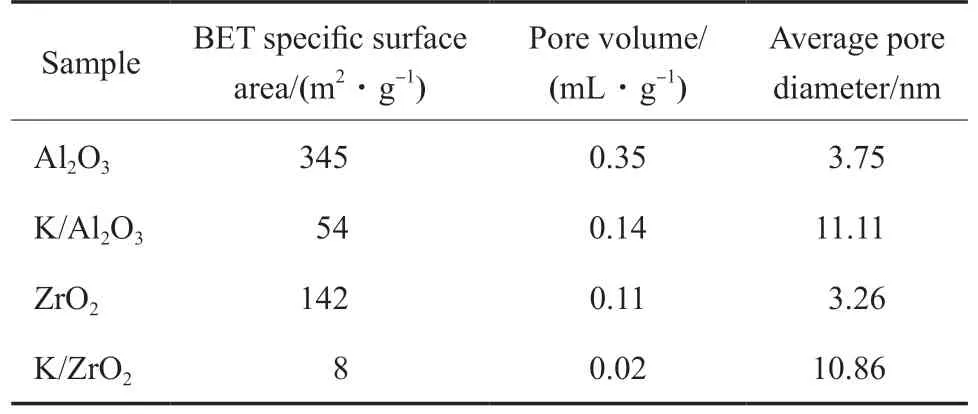
表1 載體和催化劑的織構性質Table 1 Texture properties of the supports and catalysts
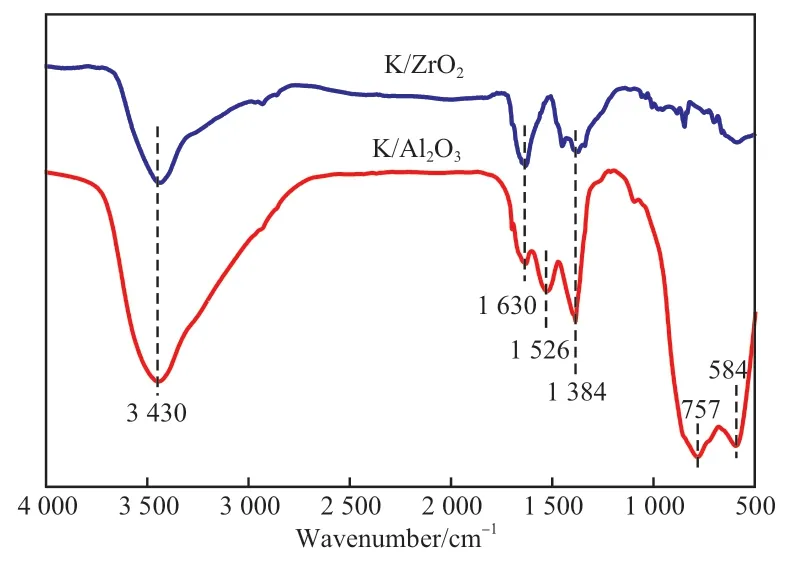
圖2 催化劑的FTIR 譜圖Fig.2 FTIR spectra of the catalysts.
2.4 TG 表征結果
圖3 為K/Al2O3和K/ZrO2催化劑的TG 曲線。由圖3 可知,K/ZrO2催化劑在整個升溫過程中失重緩慢,且失重速率較為均勻,總失重率為8.22%。35 ~115 ℃區間的失重率為2.32%,這主要是由催化劑表面吸附水的逐漸脫除引起的。115 ~260℃區間失重率為3.35%,這可歸因于部分結合水在這一階段的脫除。在260 ~600 ℃區間內,隨著溫度的不斷升高,TG 曲線失重更為緩慢,質量損失約為2.55%,可歸因于催化劑表面Zr—OH 的脫除。結合XRD 表征結果,該過程中KOH 與ZrO2產生了較強的相互作用,部分K+進入了ZrO2表面空位中形成了K—O—Zr。600 ℃以上K/ZrO2催化劑的TG 曲線基本不再發生變化。與K/ZrO2催化劑相比,K/Al2O3催化劑在整個升溫過程中持續失重且失重較為明顯,總失重率高達20.3%,遠高于K/ZrO2催化劑在相應溫度區間內的失重。200 ℃前的失重歸因于催化劑表面吸附與結合水的脫除,質量損失率為5.94%,340 ~700 ℃的質量損失率為9.25%,這是催化劑表面Al—OH 脫除造成的。可見,K/Al2O3催化劑更高的失重率表明結構中含有更多的物理吸附水和表面—OH,這與FTIR 結果一致。

圖3 催化劑的TG 曲線Fig.3 TG curves of the catalysts.
2.5 XPS 表征結果
圖4 為K/Al2O3和K/ZrO2催化劑的O1sXPS 譜圖。由圖4 可知,經分峰擬合,催化劑在528.8 ~529.2 eV,529.9 ~530.9 eV 和531.7 ~531.9 eV 范圍內均出現了3個峰,分別歸屬于表面晶格氧(Oα)、表面吸附氧(Oβ)和吸附羥基與表面吸附水中的氧物種(Oγ)[14]。與K/Al2O3催化劑相比,除吸附羥基和表面吸附水中的氧物種外,K/ZrO2催化劑上其他兩類氧物種的O 1s結合能明顯降低,表明該催化劑上氧物種的電荷密度高于K/Al2O3催化劑[15]。這是由于兩種催化劑中形成了K—O—Zr 或K—O—Al物種,前者結構中Zr4+的電負性比后者結構中Al3+的電負性小,導致與它們相鄰的氧物種電荷密度較高。此外,根據峰面積計算出Oγ占整個氧物種數量的比值Oγ/(Oα+Oβ+Oγ),K/Al2O3催化劑上Oγ所占的比例(0.19)明顯高于K/ZrO2(0.13),可見,K/Al2O3催化劑表面含有更多的吸附羥基和表面吸附水,這與FTIR 和TG 表征結果一致。
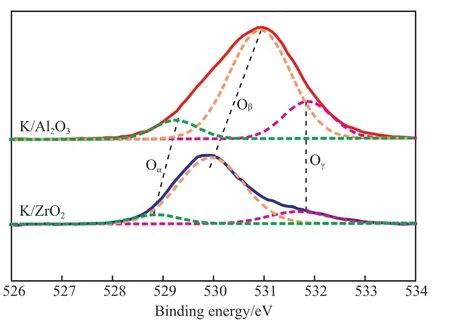
圖4 催化劑的O1s XPS 譜圖Fig.4 XPS spectra of O1s over the catalysts.
2.6 CO2-TPD 表征結果
圖5 為K/Al2O3和K/ZrO2催化劑的CO2-TPD曲線。CO2脫附溫度區間可分為3 個階段:50 ~200 ℃,200 ~400 ℃,400 ~600 ℃,各階段產生的脫附峰(Ⅰ~Ⅲ)分別對應于弱堿性、中強堿性以及強堿性位點。由圖5 可知,K/Al2O3催化劑在79 ℃和173 ℃處出現了兩個強弱不同的CO2脫附峰,均歸屬于Al2O3載體表面的弱堿性位點[16],301 ℃處的脫附峰則可歸屬于催化劑表面的中強堿性位點。K/ZrO2催化劑分別在103 ℃和192 ℃處出現了一個小的尖峰和一個不對稱的弱峰,均歸屬于載體ZrO2表面的弱堿性位點[17],在400 ~600℃之間有一個寬而強的CO2脫附峰,歸屬于催化劑表面的強堿性位點[18]。
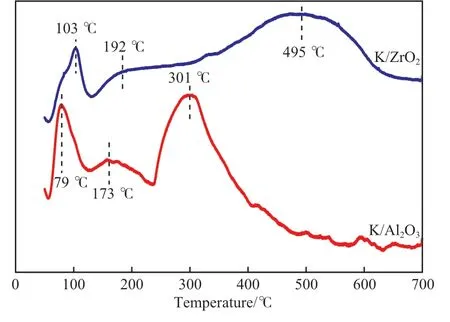
圖5 催化劑的CO2-TPD 曲線Fig.5 CO2-TPD curves of the catalysts.
進一步通過對CO2-TPD 曲線上各脫附峰面積進行定量分析,表2 給出了兩種催化劑上不同堿性位點數量和對應的堿性位密度。由表2 可知,K/Al2O3催化劑表面存在弱堿性位和中強堿性位,位點數量分別為67.4,158.3 μmol/g,對應的堿性位密度分別為1.2,2.9 μmol/(g·cm2),未出現強堿性位點。K/ZrO2催化劑表面存在弱堿性位和強堿性位,位點數量分別為64.8,160.4 μmol/g,對應的堿性位密度分別為8.1,20.1 μmol/(g·cm2),未出現明顯的中強堿性位。可見,雖然兩種催化劑上總堿性位點數量差別不大,但與K/Al2O3催化劑相比,K/ZrO2催化劑具有更高的堿強度,且表面總堿性位密度約為K/Al2O3的7 倍。結合催化劑O 1s的XPS 表征結果可知,由于Zr4+的電負性比Al3+的電負性小,導致周圍O2-上的電子云密度更高,因此K/ZrO2催化劑堿性更強,且堿性位密度更高[18]。

表2 催化劑表面不同堿性位點的數量及堿性位密度Table 2 The number and density of different basic sites on the catalyst surface
2.7 催化性能評價結果
圖6 為K/Al2O3和K/ZrO2在BDO 乙烯化反應中的催化性能。
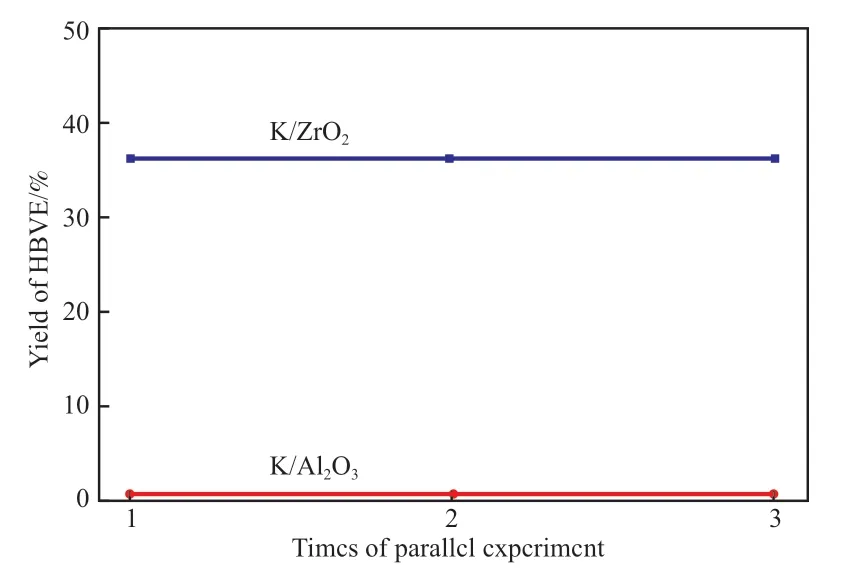
圖6 催化劑的BDO 乙烯化催化反應性能Fig.6 Catalytic performance of the catalysts for 1,4-butanediol(BDO) vinylation.Reaction conditions:m(catalyst)∶m(BDO)=0.15,150 ℃,acetylene flow rate 40 mL/min,stirring speed 400 r/min.HBVE:hydroxybutyl vinyl.
由圖6 可知,K/Al2O3催化劑上產物HBVE 收率較低,3 次平行實驗結果平均僅為0.76%;在相同反應條件下,K/ZrO2催化劑上HBVE 收率平均高達35.91%。此外,文獻[1]曾報道活性炭和4A分子篩負載KOH 催化BDO 乙烯化反應,在相近的反應條件下,HBVE 的收率分別為0.1%和1.8%。與Al2O3、活性炭以及4A 分子篩等載體相比,以ZrO2為載體負載KOH 制得的K/ZrO2催化劑對BDO 乙烯化合成HBVE 反應具有更高的催化性能。
2.8 操作參數對K/ZrO2 催化劑催化性能的影響
2.8.1 催化劑用量的影響
圖7 為催化劑用量對K/ZrO2催化劑BDO 乙烯化催化性能的影響。由圖7 可知,m(催化劑)∶m(BDO)=0.05 時,HBVE 收率僅為5.98%,這是由于催化劑用量少,催化劑的活性中心數量較少,造成HBVE 收率偏低。隨著催化劑用量的增加,活性中心的絕對數量隨之增加,HBVE 收率也呈現上升趨勢。當m(催化劑)∶m(BDO)=0.15 時,HBVE 收率為35.91%。而當m(催化劑)∶m(BDO)由0.15 進一步提高至0.25 時,HBVE 收率沒有得到明顯提高。因此,取催化劑用量m(催化劑)∶m(BDO)=0.15 最適宜。

圖7 催化劑用量對HBVE 收率的影響Fig.7 The effect of catalyst dosage on the yield of HBVE.Reaction conditions:150 ℃,acetylene flow rate 40 mL/min,stirring speed 400 r/min.
2.8.2 反應溫度的影響
圖8 為反應溫度對K/ZrO2催化劑BDO 乙烯化催化性能的影響。由圖8 可知,HBVE 選擇性隨反應溫度升高變化較大。當反應溫度為130 ℃時,BDO轉化率6.1%,HBVE 選擇性高達99.8%(HBVE 收率僅為6.09%)。隨反應溫度上升,BDO 轉化率明顯增大,但HBVE 選擇性幾乎不變。當反應溫度超過150 ℃時,BDO 轉化率增長趨勢緩慢,此時HBVE 選擇性開始驟降,導致HBVE 收率也逐漸下降。這是由于BDO 乙烯化為放熱反應,過高的反應溫度不僅不利于反應的進行,同時還導致了副反應的發生。因此,取反應溫度為150 ℃最適宜。
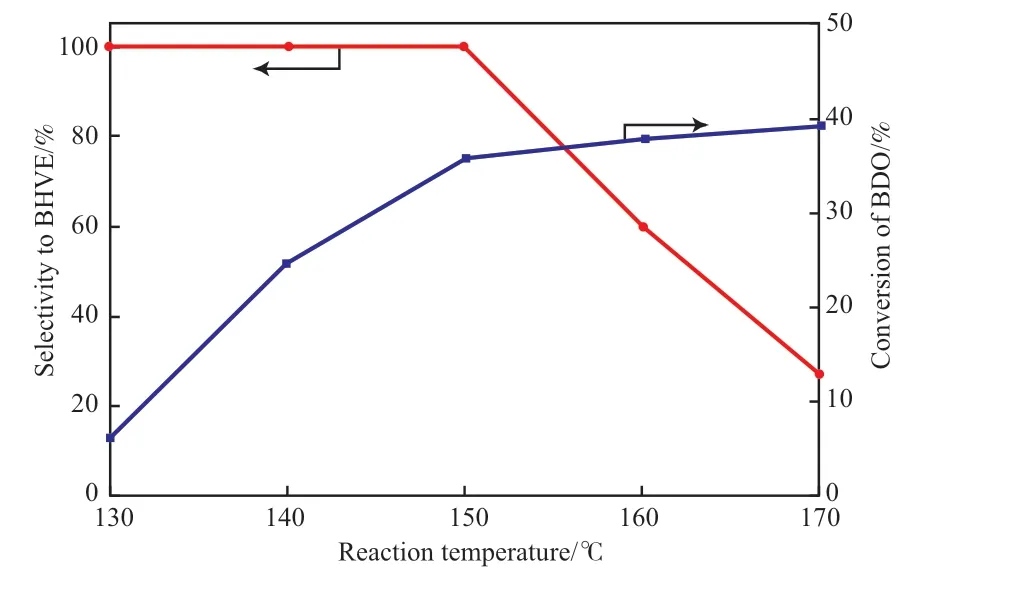
圖8 反應溫度對HBVE 選擇性的影響Fig.8 The effect of reaction temperature on the selectivity to HBVE.Reaction conditions:m(catalyst)∶m(BDO)=0.15,acetylene flow rate 40 mL/min,stirring speed 400 r/min.
2.8.3 乙炔流量的影響
圖9 為乙炔流量對K/ZrO2催化劑BDO 乙烯化催化性能的影響。由圖9 可知,當乙炔流量為10 mL/min 時,HBVE 收率低于10%,這是由于較低流量下單位時間內提供的參與反應的乙炔量較少。隨著乙炔流量的增加,HBVE 收率逐步提高,當乙炔流量為40 mL/min 時,HBVE 收率為35.91%。繼續提高乙炔流量對HBVE 收率影響甚微。因此,取乙炔流量為40 mL/min 最適宜。
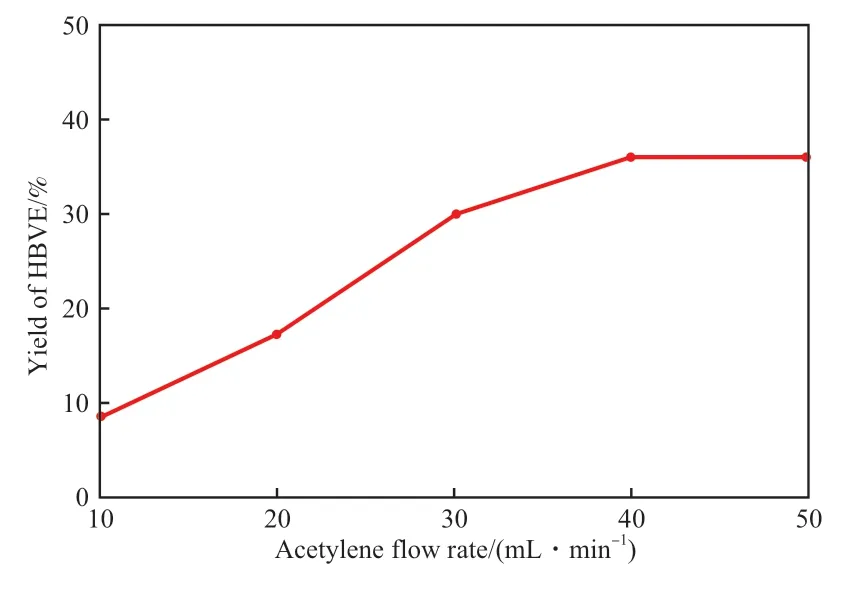
圖9 乙炔流量對HBVE 收率的影響Fig.9 The effect of acetylene flow rate on the yield of HBVE.Reaction conditions:m(catalyst)∶m(BDO)=0.15,150 ℃,stirring speed 400 r/min.
2.9 催化劑使用穩定性測試結果
圖10為K/ZrO2催化劑的穩定性。由圖10可知,新鮮K/ZrO2催化劑首次使用時,HBVE 收率較高,可達35.91%。當第2 次使用時,HBVE 收率下降至23.36%,推測原因為由于負載在ZrO2載體表面的部分K 物種與載體相互作用較弱,在首次反應過程中發生了流失。為了證實這一點,對反應濾出液進行ICP-AES 測試,結果表明K+質量濃度為7.4 mg/mL。從第2 次及此后幾次的催化性能評價結果可以看出,隨著使用次數的增加,HBVE 收率基本保持穩定,同時對每次的反應濾出液進行ICPAES 測試發現,幾乎都不含K+。當重復使用5 次后,K/ZrO2催化劑上HBVE 收率仍達21.65%,表現出較高的重復使用穩定性。
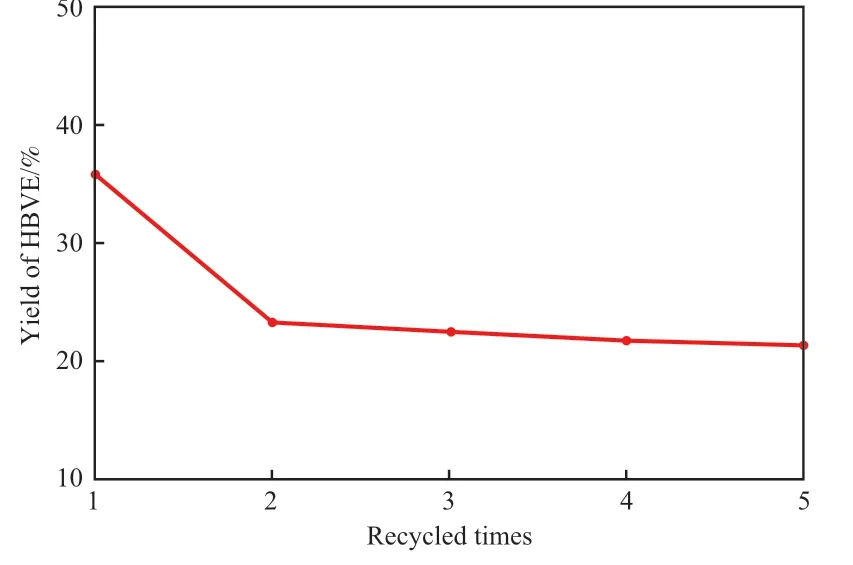
圖10 K/ZrO2 催化劑的穩定性Fig.10 The stability of the K/ZrO2 catalyst.Reaction conditions referred to Fig.6.
綜上所述,與K/Al2O3催化劑相比,K/ZrO2催化劑具有較低的比表面積和孔體積。兩種催化劑中均形成了部分K—O—Al 或K—O—Zr 結構,除了弱堿性位點外,K/ZrO2表面還存在大量的強堿性位點,而K/Al2O3催化劑表面僅存在大量的中強堿性位點,且堿性位密度遠低于K/ZrO2。結合兩種催化劑的BDO 乙烯化催化反應性能可知,對于K/Al2O3和K/ZrO2催化劑而言,比表面積不是影響催化性能的主要因素,表面堿性強弱與堿性位密度是影響該反應的決定性因素。K/ZrO2催化劑表面強堿性位點的存在以及較高的堿性位密度使其在BDO 乙烯化催化反應中表現出優異的催化性能。結合催化劑的使用穩定性測試可知,K/ZrO2催化劑不僅具有高的催化活性,還具有較優的使用穩定性,是一種性能優良的固體堿催化劑。
3 結論
1)利用沉淀法制備了Al2O3和ZrO2載體。KOH高度分散在兩種載體上,并部分形成了K—O—Al或K—O—Zr 結構。除弱堿性位點外,K/Al2O3表面僅存在中強堿性位點,而K/ZrO2表面則生成了大量的強堿性位點,表面總堿性位密度約是K/Al2O3的7 倍。
2)在相同的反應條件下,K/ZrO2催化劑的BDO 乙烯化催化活性遠高于K/Al2O3催化劑。催化劑的比表面積高低不是影響催化性能的主要因素,表面堿性強弱以及堿性位密度是決定該反應的關鍵因素。
3)KOH 的負載量為20%(w),在m(催化劑)∶m(BDO)=0.15、反應溫度150 ℃、乙炔流量40 mL/min 時,K/ZrO2催 化 劑 上HBVE 收 率 高 達35.91%。重復使用5 次后,K/ZrO2催化性能保持穩定,是一種性能優良的固體堿催化劑。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
