
甘油預處理蔗渣的木質素分離提取及結構表征
2020-11-26 09:35:16曾誠宋國杰孫海彥郭書賢孟超然孫付保
化工進展 2020年11期
關鍵詞:實驗
曾誠,宋國杰,孫海彥,郭書賢,孟超然,孫付保
(1 江南大學生物工程學院糖化學與生物技術教育部重點實驗室,江蘇無錫214122;2 中國熱帶農業科學院熱帶生物技術研究所,海南海口571101;3 南陽理工學院生物與化學工程學院河南省工業微生物資源與發酵技術重點實驗室,河南南陽473004)
在環境挑戰和能源安全的壓力下,社會正在迫切尋求可再生的碳中性資源,用于生產生物基材料、化學品和生物能源。木質素是僅次于纖維素的第二大天然高分子,也是自然界中唯一能提供可再生芳香族化合物的非石油資源。造紙工業每年要從植物中分離出大約14 億噸纖維素,同時得到5000萬噸左右的木質素副產品[1]。但目前只有不到2%的木質素被用于高附加值產品的生產,超過95%的木質素以造紙廢水的形式直接排入江河或濃縮后燒掉[2]。這種天然有機物不僅沒有得到有效利用,還造成了嚴重的污染。因此,對木質素的分離提取和結構研究顯得尤為重要。
本實驗室在前期利用甘油建立的預處理方法取得了良好的預處理效果[3-4],其中常壓甘油自催化預處理(AGO)能使甘蔗渣中的木質素脫除率達到65%以上,纖維素保留率高于95%;常壓甘油堿催化預處理(al-AGO)能使甘蔗渣中的木質素脫除75%以上,纖維素保留率高于85%。截至目前,采用甘油預處理進行木質素提取具有以下幾點優勢:①甘油木質素具有純度高、分子量小且分布窄、活性基團豐富等特點,具有較高的工業價值;②甘油熱敏性好(比熱容不足水的60%),溫度可控性高;③基質適用強,可廣泛應用于各種農林生物質原料;④屬于木質素選擇性溶解型,木質素脫除率高;⑤甘油滲透性好,它能滲透到細胞內以羥基和胞內大分子形成氫鍵,使碳水化合物保持原有結構;⑥甘油預處理過程中幾乎不產生糠醛類抑制物(<1g/kg 原料)[5]。但是,對于脫除在溶液中的木質素研究甚少。怎樣將木質素最大化提取出來,以及研究其具有哪些結構特征成為亟待解決的問題。
木質素是由苯基丙烷結構單元,即松柏醇、芥子醇和對香豆醇,經過氧化偶合形成的天然酚型高分子聚合物[6]。在進行脫木質素的過程中,木質素往往伴隨著解聚和縮合反應的發生。木質素苯基丙烷結構單元間的主要連接類型包括β-O-4'、β-β'、β-5'等[7]。另外,除了結構單元之間的連接外,木質素與碳水化合物之間還存在交聯,主要連接類型有芐基醚鍵、芐基酯鍵和苯基糖苷鍵等[8]。其中,禾本科植物原料的木質素主要通過阿魏酸與碳水化合物產生交聯[9]。
本文利用甘油對甘蔗渣分別進行AGO 和al-AGO預處理后,對溶于溶液中的木質素進行提取,分別得到自催化甘油木質素(AGOL)和堿催化甘油木質素(al-AGOL)。采用單因素實驗和正交實驗對提取方法進行優化,以期提高木質素的得率。另外,通過對木質素結構進行表征分析來研究木質素在預處理過程中所發生的結構變化。
1 實驗部分
1.1 原料與試劑
甘蔗渣(華南理工大學提供)使用前過1mm篩網,并在105℃下烘干至恒重。其主要組分含量:纖維素39.2%,半纖維素18.8%,木質素22.4%。工業甘油(純度約99.5%)購自無錫化工站。鹽酸、二氧六環、氫氧化鈉、乙酸等均購自國藥集團有限公司。
1.2 方法
1.2.1 木質素的制備
采用常壓甘油自催化預處理方法(AGO)將100g 甘蔗渣與1000g 甘油混合加熱至220℃保溫2h后,加入250mL 熱水進行冷卻,待溫度下降至80℃左右后,用G1砂芯漏斗抽濾實現固液分離,收集過濾的液體(此時濾液的甘油濃度為80%)和殘渣。在濾液中加入大量酸水(用6mol/L HCl調去離子水pH 為2,后文中的酸水都是用相同的方法配制而成),使木質素沉淀得到AGOL。此時預處理后殘渣質量為72g,殘渣組分為纖維素53.5%、半纖維素15.1%、木質素17.9%。
采用常壓甘油堿催化預處理方法(al-AGO)將100g 甘蔗渣、1000g 甘油及0.1%的NaOH 混合,在240℃下保溫10min。后面的操作與上述相同,得到木質素al-AGOL。此時預處理后殘渣質量為65g,殘渣組分為纖維素54.3%、半纖維素24.1%、木質素10.1%。
球磨甘蔗木質素(milled bagasse lignin,MBL)的獲得參照文甲龍等[10]的方法進行。在本文中MBL作為對照品用于結構表征分析的比較。
1.2.2 木質素提取的優化
(1)單因素的選擇 由于al-AGO比AGO的脫木質素效果更好,木質素提取工藝的優化建立在預處理方法al-AGO 上進行。以木質素提取率為評價指標進行單因素選擇。基礎木質素提取條件為:轉速8000r/min、離心時間為5min、甘油混合液pH為2、甘油混合液濃度為20%。選擇單因素實驗時分別考察離心轉速(2000r/min、4000r/min、6000r/min、8000r/min、10000r/min)、離心時間(3min、5min、10min、15min、20min)、甘油混合液pH(2、3、4、5、6)、甘油濃度(10%、20%、30%、40%、50%、60%)對結果的影響。每個實驗離心結束后,去除離心管中的上清液,并用清水清洗剩余固體兩次,保留離心管中的固體。將裝有固體的離心管置于105℃下烘至絕干,利用差重法,得出每個實驗所提取的木質素質量,木質素提取率的計算公式見式(1)。

(2)正交實驗優化 在單因素實驗的基礎上,設計正交實驗研究木質素工藝條件對木質素提取率的影響。以離心轉速(A)、離心時間(B)、甘油混合液pH(C)、甘油混合液濃度(D)四個因素為變量,選用L9(34)正交表設計4因素3水平正交實驗。每個實驗離心結束后,倒掉離心管中的上清液,并用清水清洗剩余固體兩次,保留離心管中的固體。將裝有固體的離心管置于105℃下烘至絕干,利用差重法計算出每個實驗所提取的木質素質量。正交實驗設計及變量水平見表1。以上每個實驗平行進行三次,實驗結果取其平均值,實驗誤差在5%以內。
1.2.3 木質素的純化
將提取得到的木質素按照固液比1∶20(g/mL)溶于90%的乙酸溶液中,再將該乙酸溶液滴入10倍體積的酸水中,再用酸水洗滌三次,離心凍干得到純化木質素[11]。將經過純化的木質素用于后續的表征分析。

表1 正交實驗設計及變量水平
1.2.4 木質素的表征
使用冷場發射掃描電子顯微鏡(SU882,日本)在放大10000倍的條件下對木質素樣品進行觀察。木質素分子量根據Li等[12]的方法進行測定:將木質素樣品(該樣品不需要進行乙酰化處理)溶解在四氫呋喃(THF)中,配制成濃度約為2.0mg/mL的溶液,并用0.45μm 的濾膜過濾后注射進樣;以THF 作為洗脫液,流速為1.0mL/min,并使用聚苯乙烯標準物質校準色譜柱,在示差折光檢測器(RID)的高效液相色譜(HPLC)Agilent 1260裝置上進行測試。采用UH-5300 型分光光度計對木質素樣品進行紫外全波長掃描,具體方法參照文獻[13]。核磁共振氫譜測定:取15mg木質素樣品溶于0.5mL 氘代二甲基亞砜(DMSO-d6)中,在60℃下進行核磁共振波譜分析(AV Ⅲ400MHz 液體核磁共振波譜儀,Bruker 公司)[14]。熱重分析:取8~10mg樣品加入熱重分析儀(DSC Q2000)中進行熱穩定分析,在N2氣氛中以10℃/min 的速率升溫,測量范圍為30~600℃[15]。抗氧化活性測定根據文獻[16]進行測量。
2 結果與討論
2.1 木質素提取工藝的優化
(1)離心轉速的選擇 離心轉速對木質素提取率的影響見圖1。由圖可見,木質素的提取率隨著離心轉速的提高而不斷增加;當轉速為8000r/min時,得到的木質素提取率達到最高值66.7%,再提高轉速對木質素提取率的影響不大。因此,實驗選擇離心轉速為8000r/min。
(2)離心時間的選擇 離心時間對木質素提取率的影響見圖2。當離心時間小于10min 時,木質素提取率隨著離心時間的延長而增加;當離心時間大于10min時,木質素增加量很小。因此,實驗選擇離心時間為10min。

圖1 離心轉速對木質素提取率的影響

圖2 離心時間對木質素提取率的影響
(3)甘油混合液pH 的選擇 文獻報道顯示,降低造紙黑液的pH 能使木質素沉淀下來。因此,本實驗考察了甘油混合溶液pH 對木質素提取率的影響。如圖3 所示,木質素的提取率隨著溶液pH 的下降而增加。當溶液的pH 為3 時,木質素提取率最大,可達到71.8%。繼續降低甘油混合液pH 至2 時,木質素提取率變化不大,反而略微減小。這是由于在木質素酸沉過程中,帶正電的木質素轉變為不溶于水的帶負電的酸析木質素。隨著pH 的降低,酸析木質素增多,依靠電性引力,酸析木質素吸附帶正電的木質素,析出顆粒逐漸增大,酸化體系的黏度增大。整個酸化體系逐漸呈黏糊狀,并可吸附大量的水分。當酸化達到一定的pH 后,大部分帶正電的木質素轉變為酸析木質素時,由于電性斥力,體系的黏度逐漸減小,酸析木質素成為松散的大小不同的顆粒狀沉淀,利于后續木質素清洗,易將雜質去除,造成木質素提取率略微下降,這與代琛等[17]的研究結果一致。因此,實驗選擇甘油混合液pH 為3。

圖3 溶液pH對木質素提取率的影響
(4)甘油混合液濃度的選擇 甘油混合液濃度對木質素提取率的影響見圖4。為了使木質素沉淀下來,最簡單的方法就是加水使甘油濃度下降,從而使不溶于水的木質素沉淀下來。由圖4可以看出,隨著加水量的增加,溶液體系中的甘油濃度不斷下降,木質素提取率也不斷增加。當溶液體系中甘油濃度為20%時,木質素提取率為69.9%,接近最大值。繼續降低甘油濃度對木質素提取率的影響不大。因此,選擇甘油濃度為20%較好。

圖4 甘油濃度對木質素提取率的影響
(5)正交實驗優化結果 在上述單因素實驗的基礎上,利用正交實驗研究工藝條件對木質素提取率的影響。以離心轉速(A)、離心時間(B)、甘油混合液pH(C)、甘油混合液濃度(D)四個因素作為考察變量,選用L9(34)正交表設計4 因素3 水平正交實驗,實驗設計及結果見表2。
根據表2中正交實驗結果及實驗結果的極差分析可知:木質素提取率影響因素的主次為C>A>D>B,較優參數組合為A3B3C2D1。木質素提取最佳工藝條件為:轉速8000r/min、離心時間15min、甘油混合液pH 為3、甘油混合液濃度10%。在此提取條件下,對使用al-AGO 預處理方法后的總濾液中的木質素進行提取,其提取率達到76%。將此種木質素提取工藝方法用于經AGO 預處理后的濾液中的木質素進行提取,其提取率達到72%。

表2 正交實驗設計及實驗結果
2.2 木質素結構表征
2.2.1 木質素的掃描電鏡
為了觀察木質素MBL、AGOL和al-AGOL的表面形態,利用SEM 對這些木質素進行表征。SEM圖像如圖5所示,這三種木質素主要呈現球形顆粒狀,這與黃陽等[18]的報道一致。AGOL 和al-AGOL與MBL 相比,在形態大小上更加均一。另外,球形木質素有疏松和親水性網絡結構的基體,具有表面積大、通透能力和水力學性能好等優點,適合于工業化、大規模的床式(固定床、移動床和流動床)吸附處理的需要[19]。后續可以將木質素用于改性,增強它的形狀穩定性,使其不易發生變形,以此來制造球形木質素吸附劑。
2.2.2 木質素樣品的元素分析
表3 給出了元素分析結構和活性官能團的數據。由表3 可知,MBL、AGOL 和al-AGOL 主要由C、H 和O 三種元素組成,并且它們的含量相差很小。有趣的是,在MBL中含有少量的N元素和S元素。有文獻顯示,這兩種元素來自于蛋白質。而在AGOL 和al-AGOL 中,S 的含量為0,可能是在甘油預處理中,由于反應比較劇烈,而S 含量又少,使得在分離純化的過程中S被除盡。

表3 木質素的元素分析
2.2.3 木質素樣品的分子量分析
木質素樣品的重均分子量(Mw)和數均分子量(Mn)通過凝膠色譜來測定。由表4得,MBL的分子量為5717g/mol,而AGOL 和al-AGOL 的分子量分別為2028g/mol和2325g/mol,MBL的分子量將近是AGOL 和al-AGOL 分子量的2倍多。這表明不論在AGO還是al-AGO預處理中,木質素大分子都會發生降解,通過解聚斷裂形成木質素片段,造成al-AGOL 和AGOL的分子量相對較小。另外,MBL的分散系數為2.05(>2),這與傳統MBL 的分散系數(>2)一致,而AGOL和al-AGOL的分散系數分別為1.47和1.46(<2)。這說明AGOL和al-AGOL相較于MBL 有著更窄的分子量分布,均一性更高。在生物精煉中,木質素的分散系數是一個重要的參數,低分散系數意味著更加好的生物化學穩定性,具有更廣泛的的應用[20]。因此,從甘蔗渣中提取得到的AGOL和al-AGOL有潛力成為一種重要的工業原料。

圖5 木質素樣品的掃描電鏡照片
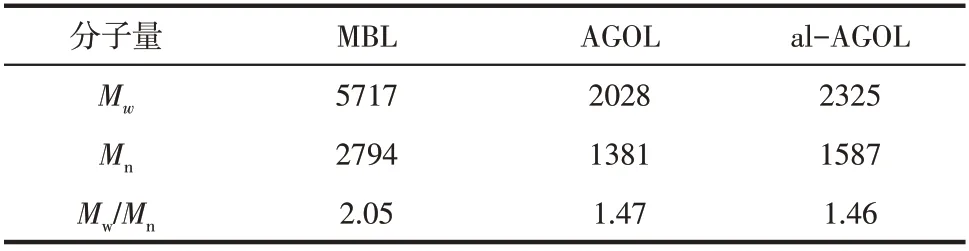
表4 木質素樣品重均分子量(Mw)、數均分子量(Mn)及其多分散度系數(Mw/Mn)
2.2.4 木質素紫外光譜分析

圖6 木質素樣品的紫外光譜圖
由圖6 所示,在木質素MBL 的紫外光譜圖中,最大吸收峰在315nm附近,說明此木質素跟其他草類木質素一樣,在側鏈含有較多的不飽和羰基。另一個吸收峰在250nm,這是由共軛雙鍵的π→π*躍遷產生的K 吸收帶[21](非封閉共軛體系的π→π*躍遷產生的特征帶)。此外,在280nm 處的肩峰是芳香族化合物B 吸收帶的特征吸收[21]。與MBL 相比,AGOL和al-AGOL 的最大吸收峰均出現在280nm左右,峰形較寬,并且有向短波長方向移動的趨勢。這可能是由于木質素經過AGO 和al-AGO 預處理后,木質素中的愈創木基較紫丁香基破壞得更多,AGOL和al-AGOL中紫丁香基單元的相對含量較高所致[22]。此外,AGOL 和al-AGOL 在315nm 處的肩峰大幅度減弱,這表明在經過甘油預處理后,木質素中酚基以及與芳香環共軛的C==C 和Cα==Cβ被大量破壞。
2.2.5 木質素樣品的1H譜分析
一維氫譜(1H NMR)核磁共振分析如圖7 所示,根據文獻將各信號峰進行歸屬[23-24],如表5 所示。在MBL譜圖中,化學位移7.2~7.5、6.8和6.2~6.4分別表示H型、G型和S型木質素芳香環上的質子信號,而β-5'結構中的Hα、β-O-4'結構中的Hβ、β-β'結構中愈創木基Hβ分別對應化學位移在5.2~5.3、4.8~4.9、4.2~4.4之間的信號峰。位于3.8處的尖銳吸收峰為甲氧基的質子吸收峰,溶劑峰DMSO出現在2.5處。此外,2.0~2.3表示甲基或者羰基上的質子,0.7~1.5是脂肪族官能團上氫原子的信號。與MBL 相比,木質素AGOL 和al-AGOL 具有相似的1H 譜吸收譜圖,但各信號峰的吸收強度發生變化。通過觀察6.2~6.8 范圍內吸收峰強度的變化,發現AGO 和al-AGO 都會破壞木質素大分子中的G型和S 型單元。尤其是在al-AGOL 的1H 譜圖中,表示G 型單元和S 型單元的信號峰減弱得更加明顯,其中表示S 型單元的信號峰沒有檢測到。另外,木質素中的主要連接結構β-5'、β-O-4'和β-β'也發生部分斷裂,其相應吸收峰的吸收強度均有所減弱。

圖7 木質素樣品的1H核磁共振譜圖

表5 木質素的1H NMR信號的主要歸屬
2.2.6 木質素的熱重分析
為了探究各木質素樣品的熱重性能,利用熱重分析儀進行了考察。如圖8所示,在熱降解的最初階段(200~350℃),MBL 的熱降解率較al-AGOL和AGOL大,說明了在木質素降解的最初階段,主要涉及β-O-4'的降解,而此時的熱降解速率主要取決于木質素中β-O-4'含量的多少。在后續的熱降解階段(350~400℃),木質素的降解速率主要取決于木質素側鏈的氧化(羰基化、羧基化和脫氫反應等)。而當降解溫度高于400℃時,主要是木質素芳環的降解,主要發生芳環飽和C—C 鍵斷裂和木質素降解為水、二氧化碳和一氧化碳等。同時,甲氧基的裂解主要發生在400~600℃。此外,al-AGOL、AGOL和MBL木質素在600℃的殘炭率分別為39.6%、40.4%和26.3%。木質素的熱穩定性可以通過木質素的最大失重率(DTGmax)來表示,al-AGOL、AGOL 和MBL 的DTGmax分 別 是388.9℃、376.6℃和365.1℃。在經過甘油預處理后,木質素的最大失重率升至高溫區域,說明了更穩定的C—C單元,如縮合單元在處理后含量增加。

圖8 木質素的熱重分析結果
2.2.7 木質素的抗氧化活性分析
為了比較各木質素的抗氧化活性大小,進行了木質素清除DPPH 自由基的實驗。如圖9 所示,木質素al-AGOL在濃度為250mg/L時抗氧化活性達到最大,此時自由基清除率為85.4%。而木質素AGOL、MBL 分別在濃度為300mg/L 和450mg/L 時達到最大值,其自由基清除率分別為83.2%和57.6%。三種木質素相比較而言,木質素al-AGOL的抗氧化活性最高,AGOL 次之,MBL 最差。另外,有研究表明,木質素添加量與抗氧化效果不總是正相關,三種木質素的自由基清除率在相應濃度達到最高值后,再提高木質素濃度,其自由基清除率均有所減小。可能是過高的木質素濃度會使得木質素間發生團聚,降低了木質素表面酚羥基與DPPH 自由基接觸的概率,使得反應活性位點減少,造成自由基清除效果變差。綜合來看,抗氧化活性最高的木質素al-AGOL 有潛力作為抗氧化劑而得到應用。
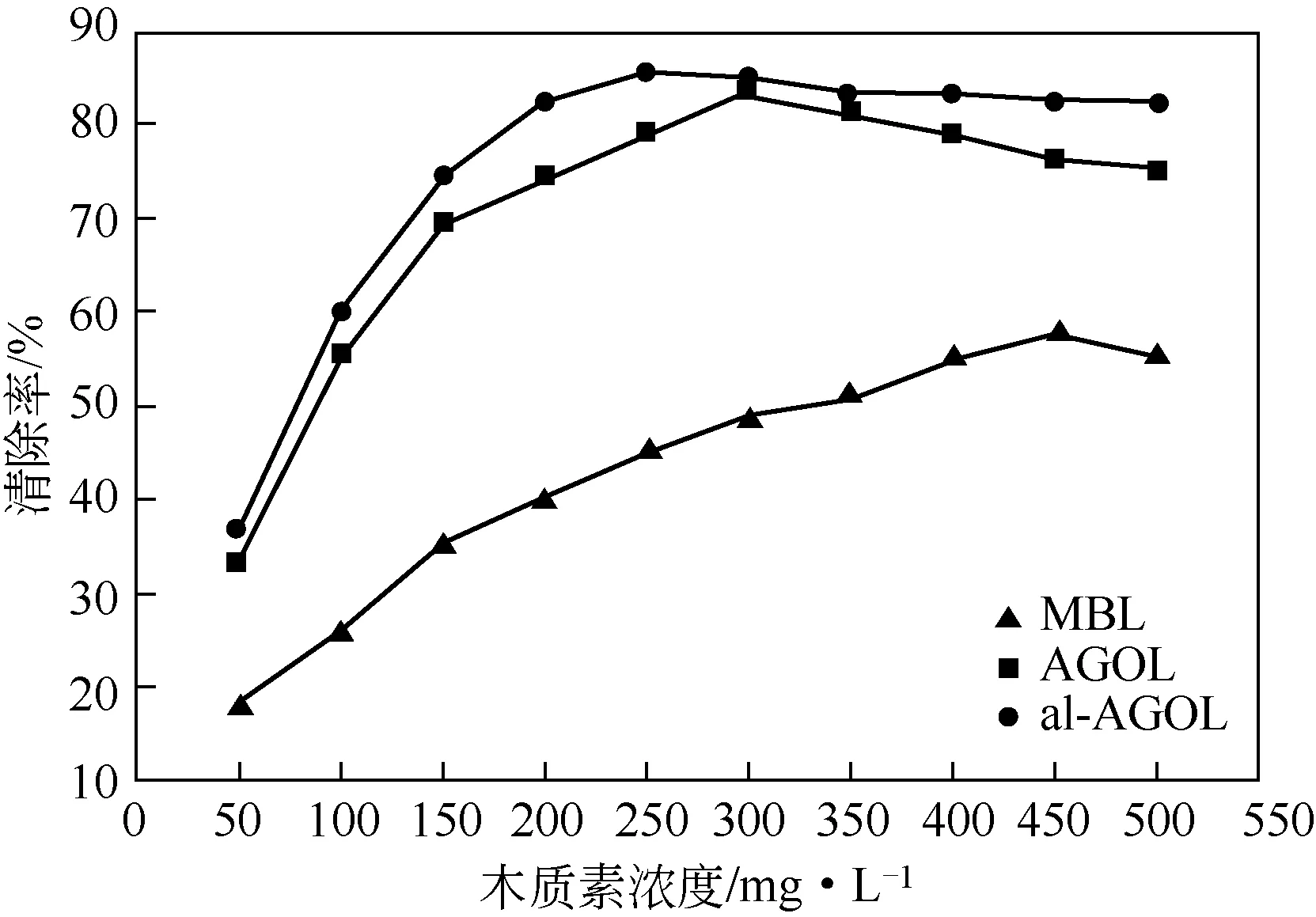
圖9 木質素的抗氧化活性分析
3 結論
(1)實驗通過單因素和正交實驗優化建立木質素最佳提取工藝為:轉速8000r/min、離心時間15min、甘油混合液pH 為3、甘油混合液濃度10%。在該條件下AGOL和al-AGOL 提取率分別達到72%和76%。
(2)各木質素樣品經過一系列表征技術分析比較,實驗發現MBL、AGOL和al-AGOL的形態均類似于球形;在AGO和al-AGO預處理過程中,木質素中的主要連接鍵β-5'、β-O-4'和β-β'均會發生斷裂,導致木質素片段從親本木質素大分子中脫離出來,造成AGOL和al-AGOL的分子量變小。熱重分析和抗氧化活性分析顯示,al-AGOL 的性能最好,AGOL次之,MBL最差。
(3)本文的研究結果為將來的木質素高值化利用提供相關理論和技術支持,具有優良特性的甘油木質素有潛力在工業應用上發揮重要作用。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
