S100A8/A9在頭頸部腫瘤發生發展中的作用及機制
2020-12-02 02:01:20冀慎英張湘豫鄒先瓊
醫學綜述 2020年21期
關鍵詞:水平
冀慎英,張湘豫,鄒先瓊
(1.桂林醫學院附屬口腔醫院口腔醫學研究所,廣西 桂林 541004; 2.桂林醫學院基礎醫學院,廣西 桂林 541100)
頭頸部腫瘤是來源于口腔、鼻腔、咽及喉等部位的惡性腫瘤的總稱,常見類型包括口腔鱗狀細胞癌(oral squamous cell carcinoma,OSCC)、口咽鱗狀細胞癌(oropharyngeal squamous cell carcinoma,OPSCC)、食管鱗狀細胞癌(esophageal squamous cell carcinoma,ESCC)、鼻咽鱗狀細胞癌(nasopharyngeal squamous cell carcinoma,NSCC)、喉鱗狀細胞癌(laryngeal squamous cell carcinoma LSCC)、甲狀腺癌(thyroid cancer,TC)等[1-2]。OSCC是最常見的頭頸部鱗狀細胞癌(head and neck squamous cell carcinoma,HNSCC),常伴有口腔癌前病變,如口腔白斑等[3]。隨著研究的深入發現,S100A8/A9的異常表達與頭頸部腫瘤的發生發展密切相關[1]。
1 S100A8/A9的結構與功能
S100A8和S100A9均屬于S100蛋白家族,分子量分別為10 800和13 200[4],其分子結構均由兩個不同的螺旋-環-螺旋配基組成,兩側為疏水區,中央為鉸鏈區,鉸鏈區與其他靶蛋白結合[4]。S100A8及S100A9均為酸性鈣離子結合蛋白,兩者以鈣離子依賴性方式形成異源二聚體S100A8/A9(即鈣防衛蛋白)[5]。S100A8/A9能誘導中性粒細胞趨化和黏附,促進白細胞花生四烯酸的轉運和代謝,調節吞噬細胞遷移和中性粒細胞還原型煙酰胺腺嘌呤二核苷酸磷酸氧化酶活化過程中微管蛋白依賴性細胞骨架的代謝,具有促炎、抗菌、抗氧化和誘導凋亡等功能[5]。
S100A8/A9以組織特異性方式表達,在多種腫瘤的發生發展過程中發揮重要作用[1]。S100A8/A9在皮膚癌、乳腺癌、前列腺癌、卵巢癌、胰腺癌、結直腸癌、膀胱癌、胃癌[6]以及部分TC組織[1]中高表達,具有促進腫瘤發生發展的作用[1]。S100A8/A9在OSCC、OPSCC、ESCC、NSCC及LSCC等頭頸部腫瘤組織中低表達,一般認為發揮了抗腫瘤作用[1]。S100A8/A9在不同腫瘤組織中的作用可能取決于其在細胞內外的水平和位置[5]:其在細胞外高水平時能誘導細胞凋亡;細胞內高水平時可調節上皮細胞間質化與間質細胞上皮化之間的信號級聯,誘導癌細胞侵襲能力降低[5]。
2 S100A8/A9在頭頸部腫瘤發生發展中的作用
2.1S100A8/A9與OSCC和OPSCC S100A8/A9表達失調是口腔和口咽腫瘤發生的一個特征[1]。在OSCC、OPSCC細胞中S100A8/A9的表達水平較口腔和口咽非腫瘤性復層鱗狀上皮顯著降低[1]。S100A8/A9的表達在高分化、中等分化、低分化以及非角質化基底細胞樣癌HNSCC中逐漸降低,提示S100A8/A9的表達水平在HNSCC的發生發展中逐漸下調[7]。一般認為OSCC中S100A8/A9表達的下調與腫瘤分化不良[1]以及DNA甲基化增加[8]有關。
S100A8/A9可能調節OSCC細胞的惡性特征[1]。在高分化的人OSCC細胞系中,采用短發夾RNA沉默內源性S100A8/A9后,細胞基質金屬蛋白酶(matrix metalloproteinase,MMP)-2的活性及細胞遷移和侵襲能力增強[9];相反,當S100A8/A9在KB細胞(S100A8/A9陰性表達的細胞)過表達后,MMP-2的表達、細胞遷移及侵襲能力減弱[9],表明S100A8/A9能夠負性調控MMP-2蛋白的產生和相關酶的活性。另一方面,在MC38結腸癌細胞和LLC肺癌細胞中,S100A8/A9的下調顯著降低了MMP-2和MMP-9在癌細胞中的表達,進一步導致癌細胞遷移和侵襲能力降低[10],說明基質來源和腫瘤來源的S100A8/A9對于調節MMP-2和MMP-9的表達有功能差異。用單核/巨噬細胞條件培養基誘導MC38和LLC細胞表達S100A8/A9,與未表達S100A8/A9的MC38和LLC癌細胞相比,細胞內鈣離子水平降低[9]。當誘導的S100A8/A9被敲除時,癌細胞中鈣離子水平降低的現象被消除[9],提示S100A8/A9可通過調節癌細胞內鈣離子的水平調節MMP-2、MMP-9的表達[9]。使用小鼠異種移植腫瘤模型發現,KB細胞和KB-EGFP細胞形成的腫瘤明顯大于KB-S100A8/A9細胞[8],說明S100A8/A9與控制細胞發育和分化、細胞間信號和相互作用以及細胞形態的基因網絡相互作用,下調了與侵襲和腫瘤發生相關的基因的表達[8]。
研究發現,S100A8/A9在OSCC中表達的下調與G2/M期細胞周期阻滯密切相關[11]。典型的G2/M細胞周期檢查點信號通路受細胞周期檢測點激酶1(checkpoint kinase 1,Chk1)的控制[11]。細胞周期中的細胞分裂周期蛋白25同源蛋白C(cell division cyclin 25 homolog C,Cdc25C)是激活細胞周期蛋白B1(cyclin B1)/細胞周期蛋白依賴激酶1復合物和進入有絲分裂所必需的[11]。S100A8/A9的表達增加了Chk1(Ser345)的磷酸化和蛋白磷酸酶2A的活性,p-Chk1(Ser345)使Cdc25C在Ser216處磷酸化后與分子伴侶14-3-3β結合,形成無活性的p-Cdc25C/14-3-3b復合物,導致Cdc25C失活并在胞質中積聚[11]。另一方面,p-Cdc25c(Thr48)可使p-Cdc2(Thr14/Tyr15)去磷酸化成為有活性的Cdc2,Cdc2與cyclin B1結合成為cyclin B1/Cdc2復合物,從而進行有絲分裂[11]。但Cdc25C的Thr48和Ser216殘基不能同時磷酸化[11]。當蛋白磷酸酶2A活性增強后可使p-Cdc25c(Thr48)去磷酸化,導致p-Cdc2(Thr14/Tyr15)積聚,cyclin B1表達下調,細胞周期在G2/M檢查點停止,導致細胞周期阻滯[11](圖1A)。
表皮生長因子受體(epidermal growth factor receptor,EGFR)在OSCC、OPSCC中高表達[7]。EGFR可被胱天蛋白酶(caspase)1、3和7水解和翻譯后修飾,故細胞表面EGFR的變異可能反映caspases-1、caspases-3和caspases-7的裂解[7]。S100A8/A9在頭頸部腫瘤細胞中過表達時,caspase-1、caspases-3和caspases-7的轉錄水平和活性提高,導致EGFR水解作用增強,EGFR水平降低,進而影響EGFR依賴的下游信號轉導[7]。在TR146細胞沉默S100A8/A9后,細胞caspase-3和caspase-7的活性顯著降低,而EGFR水平升高,這說明S100A8/A9的表達與EGFR蛋白呈負相關[7](圖1B)。該研究也表明S100A8/A9相關的EGFR下調可能有助于S100A8/A9高表達的HNSCC患者總生存率的提高[7]。

OSCC:口腔鱗狀細胞癌;p-Chk1(Ser345):在絲氨酸345處磷酸化的周期檢測點激酶1;Cdc25C:細胞分裂周期蛋白25同源蛋白C; p-Chk1(Ser216):在絲氨酸216處磷酸化的周期檢測點激酶1;14-3-3b:分子伴侶;PP2A:蛋白磷酸酶2A;p-Chk1(Thr48):在蘇氨酸48處磷酸化的周期檢測點激酶1;cyclin B1:細胞周期蛋白B1;p-Cdc2(Thr14/Tyr15):在蘇氨酸48處或酪氨酸15處磷酸化的細胞周期蛋白依賴激酶1; EGFR:表皮生長因子受體;caspase:胱天蛋白酶
在人乳頭瘤病毒(human papillomavirus,HPV)陰性、S100A8/A9高表達的OSCC、OPSCC細胞中,S100A8/A9減弱了癌細胞的惡性表型,起到了抑癌作用[7]。S100A8/A9可能通過調節酪蛋白激酶Ⅱ介導的體外HPV E7蛋白的磷酸化而抑制病毒的致癌活性[12]。HPV誘導的S100A8/A9的間接下調可能是一種額外的致瘤機制[7]。
2.2S100A8/A9與ESCC 在ESCC中S100A8/A9表達下調,且其表達水平與ESCC的分化程度呈負相關[13]。低分化ESCC組織細胞與中度和分化良好的ESCC組織細胞相比,S100A8/A9的表達顯著下調,甚至不表達[13],這可能與S100A8/A9在食管鱗狀上皮細胞中能阻滯細胞周期進程,進而抑制細胞生長有關[13]。
4-硝基喹啉-1-氧化物(4-nitroquinoline-1-oxide,4-NQO)是一種前體致癌劑,在舌部黏膜中含量較高,故4-NQO也可作為舌癌的標志物之一[14]。環加氧酶2(cyclooxygenase-2,COX-2)是合成前列腺素的關鍵酶,也是核因子κB(nuclear factor-kappaB,NF-κB)的下游靶點[14]。經4-NQO處理的缺鋅性小鼠的ESCC中S100A8的免疫反應顯著增強[14]。當4-NQO局部誘導缺鋅COX-2-/-小鼠發生食管癌時,S100A8/A9在近端胃癌/舌癌中的表達上調[14]。免疫組織化學顯示,S100A8/A9、晚期糖基化終末產物受體(receptor for advanced glycosylation end product,RAGE)、NF-κB、p65和cyclin D1在缺鋅性小鼠近端胃或舌的癌前病變和癌中共表達,提示RAGE-S100A8/A9炎癥信號通路在近端胃/舌的癌前病變和癌變中被激活[14]。因此,缺鋅可能調節S100A8/A9的表達,并調節S100A8/A9與RAGE的相互作用和S100A8/A9與下游NF-κB/COX-2信號通路間的聯系,從而促進食管細胞增殖和癌變[14](圖2A)。通過特殊飲食建立缺鋅性增生和補充鋅的大鼠食管組織模型發現,S100A8/A9在缺鋅性增生食管中的表達與補鋅組織相比顯著增強,表明補鋅能恢復S100A8/A9基因在體內的表達和食管的生理表型[15]。缺鋅性小鼠ESCC的NF-κB通路還受微RNA(microRNA,miRNA)的調控[16]。miR-31是最常見的腫瘤相關失調的miRNA之一,在ESCC、舌鱗狀細胞癌等HNSCC組織中高表達[17]。缺鋅性小鼠ESCC誘導的癌基因miR-31的過度表達能夠下調絲氨酸/蘇氨酸激酶40(Stk40),進而通過S100A8/A9激活NF-κB和RAGE,促進炎癥發生和細胞增殖,產生食管癌前表型(圖2B)[17]。以上研究表明,鋅缺乏激活了S100A8/A9-RAGE/NF-κB和miR-31/Stk40/NF-κB信號通路,促進細胞增殖,抑制細胞凋亡,從而促進食管癌的發生發展[17]。
2.3S100A8/A9與NSCC及LSCC 在NSCC組織中,S100A8/A9的表達水平顯著低于癌旁正常組織[18]。S100A8/A9在鼻咽惡性腫瘤的上皮中表達不明顯,但在非腫瘤性上皮的淺層表達[18]。S100A9在癌細胞和正常咽上皮細胞中的免疫反應性均為陰性,但在所有NSCC的間質炎癥細胞中均有免疫反應性[19]。在更晚期NSCC區域淋巴結轉移增加中,S100A9的表達在炎癥腫瘤微環境中上調,但在癌細胞未上調[19]。S100A8/A9通過劑量效應影響鼻咽癌細胞的增殖與凋亡[20],低水平S100A8/A9(S100A8/A9<10 mg/L)能夠促進鼻咽癌細胞株CNE1及CNE2的增殖,而S100A8/A9水平超過30 mg/L則會抑制細胞生長[21]。
在LSCC組織中,S100A9的表達水平顯著低于癌旁正常組織[22]。S100A8的3′非翻譯區攜帶一個直接與miR-24結合的特異位點,在LSCC的Hep2細胞中miR-24能在翻譯水平直接靶向S100A8,顯著誘導Hep2細胞的形態學改變,并在阻斷S100A8蛋白后顯著抑制Hep2細胞的增殖和侵襲[23];另一方面,circMAN2B2可通過抑制miR-1205促進S100A8的表達,進而促進細胞的增殖、侵襲和遷移[24]。
2.4S100A8/A9與TC TC是來源于內分泌器官的最常見的惡性腫瘤之一,濾泡狀癌和乳頭狀癌是其最常見的兩種類型,且均不表達S100A9[25]。S100A9在TC中的陽性率普遍較低,但未分化癌除外[25]。在未分化癌中,S100A9呈較高表達[25]。一般認為,升高的S100A9蛋白可能會導致TC的去分化。
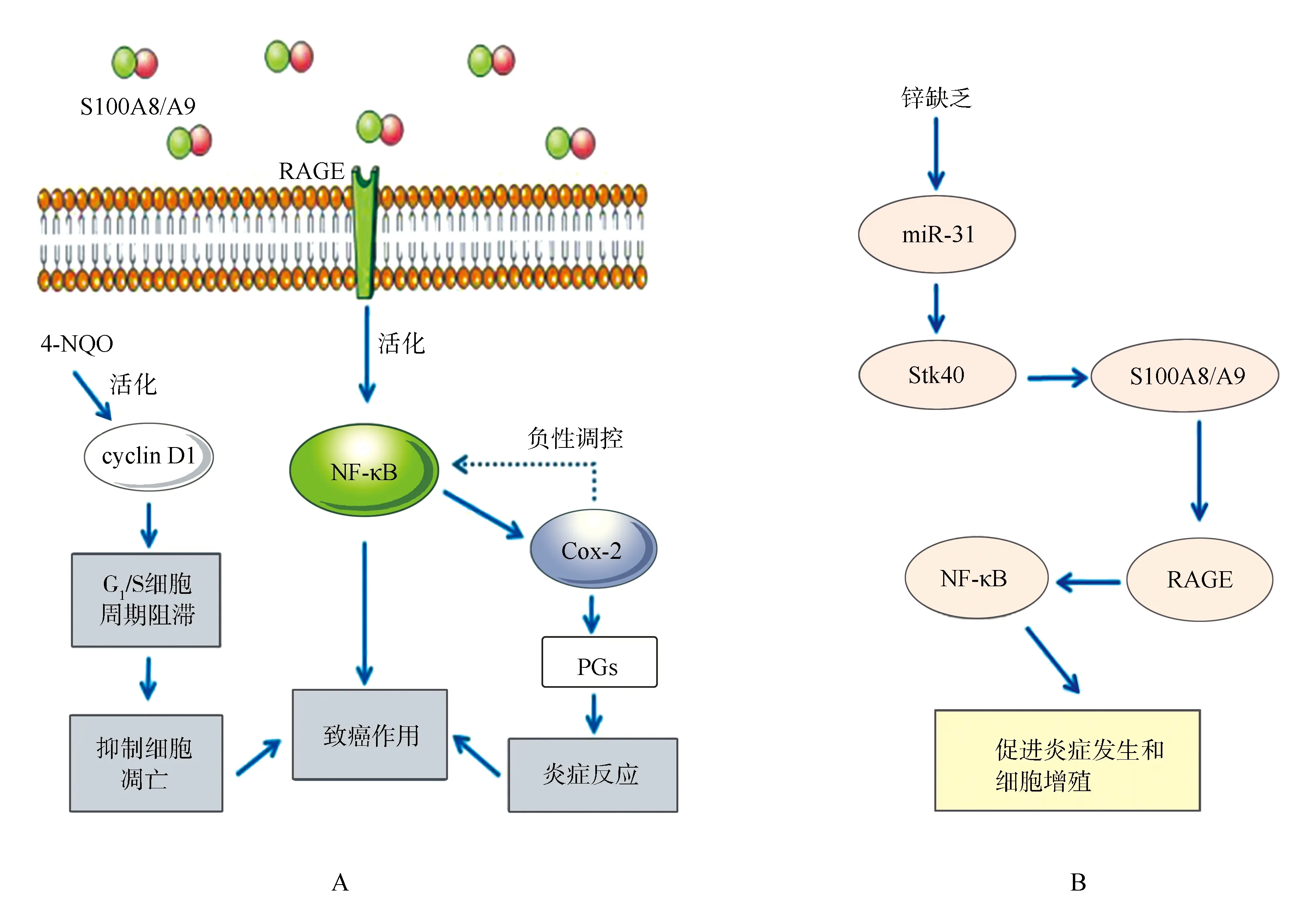
ESCC:食管鱗狀細胞癌;RAGE:晚期糖基化終末產物受體;4-NQO:4-硝基喹啉-1-氧化物;cyclin D1:細胞周期蛋白D1;NF-κB:核因子κB;COX-2:環加氧酶2;PGs:前列腺素;miR-31:微RNA-31;Stk40:絲氨酸/蘇氨酸激酶40
間變性甲狀腺癌(anaplastic thyroid carcinoma,ATC)是TC中最惡性的類型,其特點是早期轉移和局部器官浸潤[26]。在ATC中,S100A8/A9的表達高于非腫瘤性甲狀腺組織和高分化甲狀腺腫瘤[27]。S100A8通過與RAGE相互作用激活下游的p38、胞外信號調節激酶1/2(extracellular regulated kinase 1/2,ERK1/2)、c-Jun氨基端激酶(c-Jun N-terminal kinase,JNK)、促分裂原活化的蛋白激酶(mitogen activated protein kinase,MAPK)通路,進而刺激腫瘤細胞生長[27]。通過短發夾技術敲除內源性S100A8,抑制S100A8介導的ATC的體外增殖和體內腫瘤形成,提高動物存活率[27]。S100A8表達的下調能抑制蛋白激酶B的磷酸化,誘導細胞色素C、caspase-3及caspase-9等促凋亡基因表達,進而誘導細胞凋亡[28]。因此,S100A8的靶向性可能有利于ATC患者的預后及治療[27]。
甲狀腺乳頭狀癌(papillary thyroid carcinoma,PTC)是最常見的TC,預后良好[29]。PTC晚期分泌大量與腫瘤相關的巨噬細胞,這些巨噬細胞是S100A8/A9的主要來源,所以PTC患者的S100A8/A9水平顯著升高[6]。氧化應激可導致活性氧類的過度產生或清除不足,這些高活性代謝物通過刺激過氧亞硝酸鹽和其他致突變劑導致DNA損傷[6]。S100A8/A9可通過S100A9 C端的HHH結構域與花生四烯酸結合,S100A9在Thr113處的磷酸化可激活還原型煙酰胺腺嘌呤二核苷酸磷酸氧化酶產生活性氧類,并激活NF-κB信號通路[1]。有研究表明,PTC患者中的氧化應激指數、血清脂質過氧化物及S100A8/A9水平均呈上升趨勢,在甲狀腺全切除術后血清S100A8/A9的水平明顯下降[6]。提示S100A8/A9可能是這類甲狀腺惡性腫瘤的一個有用的生物標志物[6]。另有研究顯示,與健康對照組相比,良性甲狀腺腫和PTC患者的尿蛋白中S100A8和S100A9蛋白的表達均顯著降低[29]。
3 S100A8/A9基因的甲基化與頭頸部腫瘤
DNA甲基化、組蛋白修飾等表觀遺傳改變可在整個細胞生命周期中持續,并遺傳給子代細胞[30]。在腫瘤細胞中,常存在正常DNA甲基化模式的異常改變,表現為全基因組廣泛低甲基化及抑癌基因CpG島區域高甲基化[31]。在許多腫瘤中,啟動子甲基化導致的基因沉默可能是癌變過程的早期事件,可能較突變和缺失導致的基因結構失活更常見[3]。研究表明,OSCC患者遠端正常黏膜組織的平均甲基化水平顯著低于OSCC區的甲基化水平,同時顯著高于健康供者正常黏膜的甲基化水平[3]。在HNSCC中S100A9表達的下調與增強的甲基化水平呈負相關,而HNSCC中S100A8表達的下調與增強的甲基化可能并不相關[8]。
4 展 望
S100A8/A9在頭頸部腫瘤發生發展中發揮了重要作用,有可能成為頭頸部腫瘤預防、治療和預后評定的新指標[1,5-9]。S100A8/A9在頭頸部腫瘤發生發展中的作用機制與G2/M期細胞周期阻滯、EGFR異常、RAGE/NF-κB信號通路等密切相關。近年來關于表觀遺傳調控通過影響S100A8/A9的表達,進而影響頭頸部腫瘤發生發展進程的研究逐漸增多[1,30],S100A9在HNSCC中的低表達與增強的DNA甲基化水平呈負相關[8]。隨著對S100A8/A9在頭頸部腫瘤發生發展中作用機制研究的深入,表觀遺傳調控在頭頸部腫瘤發生發展中的作用也將成為研究的熱點。相信隨著S100A8/A9的作用及調控機制研究的不斷深入,S100A8/A9將會在頭頸部腫瘤的預防、診斷及治療等方面發揮更加重要的作用。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
