禾本科主要農作物葉綠體基因組研究進展
2020-12-04 08:07:08李裕華任永康趙興華劉江韓斌王長彪唐朝暉
生物技術通報 2020年11期
李裕華 任永康 趙興華 劉江 韓斌 王長彪 唐朝暉
(1.山西大學生物工程學院,太原 030006;2.山西省農業科學院作物科學研究所,太原030031;3.山西省農業科學院生物技術研究中心,太原 030031;4.山西省農業科學院,太原 030031)
作為光合作用的主要場所,葉綠體被視為推進早期生命進化的能量起源。此外,葉綠體在植物生理學和發育的其他方面也起著至關重要的作用[1],包括氨基酸、核苷酸、脂肪酸、植物激素、維生素和大量代謝產物的合成以及硫和氮的同化等重要的生理生化活動[2]。葉綠體基因組研究不僅有助于通過遺傳轉化體系改良葉綠體功能和選育新品種,而且有利于增強對植物生物學和生物多樣性的理解。全面了解葉綠體基因組及其在生物進化中的作用,能夠為植物科學的系統發育研究提供關鍵信息,同時也為探索核基因組、線粒體基因組以及葉綠體基因組3個基因組之間的關系提供新的思路。
禾本科作物是人類糧食和牲畜飼料的主要來源,如小麥、水稻、玉米、大麥及高粱等。研究發現,與其他植物相比,禾本科作物葉綠體基因組在進化過程中的速度加快[3],同時結構上發生了一系列變化[4],因此,禾本科植物為葉綠體基因組的進化研究提供了很好的條件。
1 禾本科作物葉綠體基因組的特征
1962年,通過電子顯微鏡觀察衣藻(Chlamydomonas)葉綠體,發現了DNA纖絲,由此得出葉綠體內存在DNA分子[5]。此后,在很多植物中先后發現葉綠體DNA。1986年,植物葉綠體基因組——煙草(Nicotiana tabacum)全序列最先被發表[6],同年Ohyama又測得地錢(Marchantia polymorpha)葉綠體基因組的完整序列[7]。隨后,葉綠體基因組數據庫迅速增加充實。
1.1 主要禾本科作物葉綠體基因組序列特征
目前,在NCBI(National Center for Biotechnology Information)上公布的禾本科葉綠體完整基因組序列有1 219個,其中稻屬(Oryza L.)有104個,玉米屬(Zea L.)有9個,小麥屬(Triticum L.)有41個。小麥(Triticum aestivum)[8]、水稻(Oryza sativa)[9]、玉米(Zea mays)[10]、大麥(Hordeum vulgare)、高粱(Sorghum bicolor)[11]、黑麥(Secale cereale)[12]等主要禾本科作物的葉綠體全基因組序列也已隨之公開(表1)。
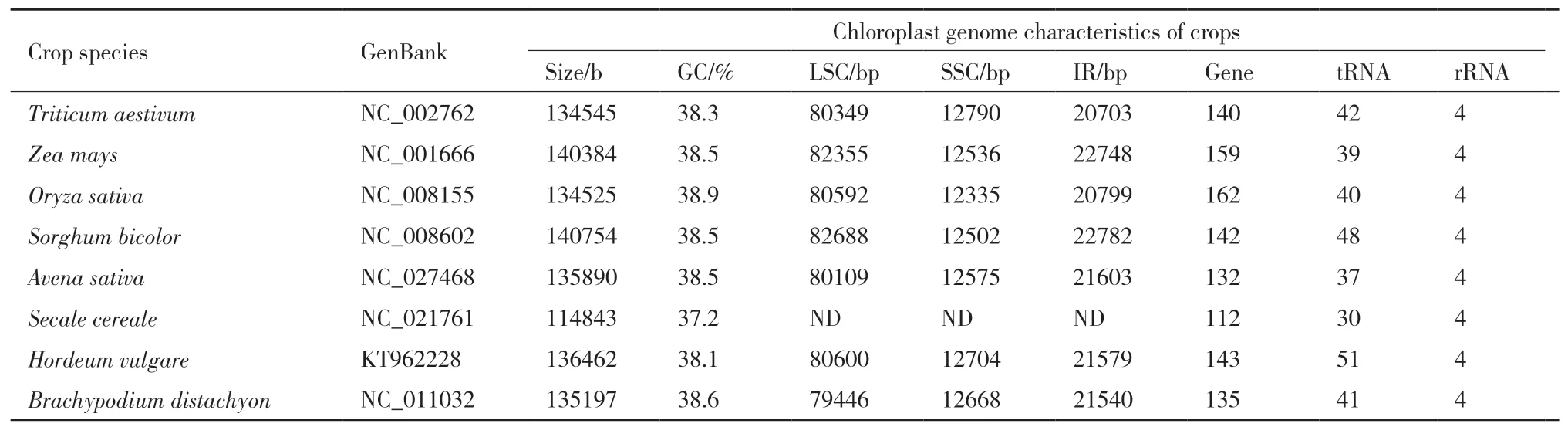
表1 完成測序的部分禾本科作物葉綠體基因組序列分析
從這些完成測序的主要禾本科作物葉綠體基因組的大小來看,一般在115 kb-150 kb[13-14],表1中最大的是高粱(140 752 bp),最小的是黑麥(114 843 bp),小麥、水稻、燕麥、大麥、二穗短柄草之間葉綠體基因組大小相差不多。禾本科作物葉綠體基因組中GC含量均在37.1%(黑麥)-38.9%(水稻)。LSC區的長度約80 kb,SSC區的長度約13 kb,IR區的長度約20 kb[15]。水稻葉綠體基因組中基因數量最多,為162個,小麥、高粱、燕麥、大麥、二穗短柄草之間葉綠體基因組中基因數目差異不大。tRNA在各物種之間數目和種類存在差異,但rRNA的數目和種類在各物種之間保持穩定。
1.2 主要禾本科作物葉綠體基因組結構特征
禾本科作物葉綠體基因組一般為共價閉合環狀DNA,在細胞中以多拷貝的形式存在[13]。除環狀外,有極少數植物的葉綠體基因組為多環型或線型,如雙鞭甲藻(Crypthecodinium cohnii)的葉綠體基因組為多環型結構[16],傘藻(Acetabularia)的葉綠體基因組為線型結構[17]。以禾本科模式作物水稻為例(圖1),從NCBI上下載水稻葉綠體全基因組序列(登錄號為NC_031333.1),并通過OGDRAW(http://chlorobox.mipmpgolm.mpg.de/OGDraw.html)繪制工具繪制。在圖1中可以看出,葉綠體基因組由4個基本部分組成,分別是大單拷貝區(LSC),小單拷貝區(SSC),反向重復區A(IRA)和反向重復區B(IRB),2個片段的反向重復序列被大單拷貝和小單拷貝所隔開,2個IR區域的序列相同,但方向相反。葉綠體基因組上存在高的基因轉換能力[18],確保了2個IR序列的一致與穩定。禾本科植物在進化過程中,反向重復區是葉綠體基因組進化過程中延展或縮小的區域。基因組在進化的過程中,IR區序列邊緣區也發生了變化[19],隨著IR邊界的擴張與收縮,有些基因進入IR區,有些基因進入單拷貝區,導致不同物種間的基因數量發生變化。
因此,葉綠體基因組的大小變化在進化過程中主要受到反向重復區的長度變異所影響[20]。禾本科作物在進化過程中,其葉綠體基因組的結構發生了變化[4,19]。在禾本科作物與煙草的葉綠體基因組比對時發現,基因排列的順序及轉錄方向存在差異。禾本科作物葉綠體基因組中基因的排列從trnR至trnfM、trnG至psbD以及trnT區域內出現了3次倒置[19,21]。其他物種中暫未發現此種現象,說明倒置發生在禾本科植物的共同祖先中[4,19,22]。
2 禾本科葉綠體基因組中的基因類型分布

圖1 水稻(Oryza sativa)葉綠體基因組結構圖
禾本科植物葉綠體基因組含有許多功能基因,大約編碼110-130個基因[13],主要分為4大類:第一類是與光合作用有關的基因,包括光系統Ⅰ類基因、光系統Ⅱ類基因、NAD(P)H脫氫酶類基因、細胞色素b/f復合體類基因、ATP合成酶類基因、Rubisco大亞基類基因,這些基因散布在LSC區域;第二類是與自我復制相關的基因,包括核糖體RNA類基因、轉運RNA類基因、RNA聚合酶類基因、編碼核糖體大亞基蛋白類基因、編碼RNA聚合酶類的亞基基因、編碼核糖體小亞基蛋白類基因;第三類是參與葉綠體中生物合成有關的基因,包括成熟酶基因、乙酰輔酶A羧化酶基因、蛋白酶基因、包膜蛋白基因、細胞色素C合成基因和翻譯起始因子基因等;第四類為開放式閱讀框(Open reading frame,ORF)即一些功能未知的基因。ycf3和ycf4的產物充當光系統Ⅰ復合物的裝配因子[23-26],Wicke建議將這兩個基因重命名為pafI和pafII,即光系統Ⅰ的組裝因子I和II[15,26],所以表中將這個基因劃分到編碼光系統Ⅰ的基因中(表2)。
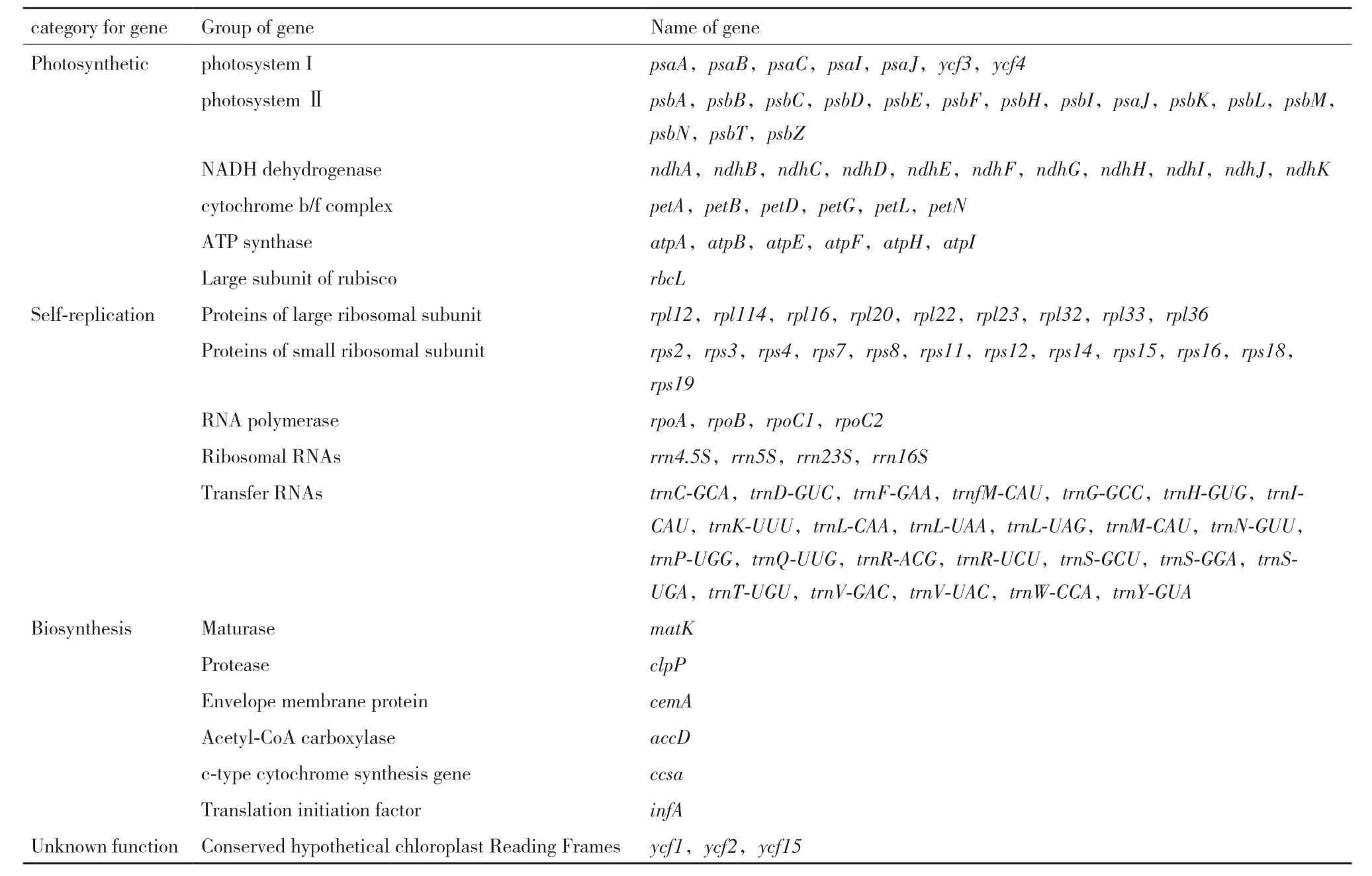
表2 葉綠體基因組中的基因類型分布
3 禾本科葉綠體基因組中的基因序列解析
隨著禾本科作物的葉綠體基因組測序數據的不斷公布,與基因序列相關的工作已經逐步開展,通過基因序列比對,進一步從基因缺失退化、RNA編輯位點預測等方面進行研究,為系統發育組學的研究奠定了基礎。
3.1 禾本科葉綠體基因組中的基因缺失退化現象
禾本科植物葉綠體基因組的基因差異較少,參與編碼光系統Ⅰ、光系統Ⅱ、NAD(P)H脫氫酶、細胞色素b/f復合體、ATP合成酶、核糖體RNA、轉運RNA、RNA聚合酶、編碼核糖體蛋白的基因,以及參與葉綠體生物合成的基因都是高度保守的。但是,在不同作物中一些基因在進化過程中存在退化缺失現象。如在禾本科植物中ycf1和ycf2序列發生了逐漸退化缺失[27]。小麥、玉米、大麥、高粱及黑麥中的accD序列逐漸退化缺失[19],但水稻中仍然存在。ycf2和ycf15序列在黑麥中依然存在,但在小麥、玉米、水稻、大麥赫爾高粱中都已缺失。ycf15序列由于其高度保守且含有豐富的變異位點[28],使得ycf15在系統發育學和分子標記領域存在巨大的研究空間和潛力[29-30],但在禾本科植物中由于ycf15結構變異大,存在3種基因結構,限制了其發展[8,31]。葉綠體基因組中還存在內含子丟失的情況,禾本科植物最初分化產生的物種發生clpP內含子的丟失,隨后又發生了rpoC1內含子丟失[19]。
3.2 禾本科作物葉綠體基因組RNA編輯
作為高等植物葉綠體基因轉錄后表達調控的一種重要方式,RNA編輯是指在基因轉錄后mRNA中發生的核苷酸堿基的插入、缺失或替換,導致核苷酸序列的改變,從而改變原來遺傳信息的過程,導致同一基因翻譯出多種氨基酸序列不同的蛋白質[32-34]。RNA編輯一般通過改變蛋白質一級結構中氨基酸的組成最終影響蛋白質的功能。RNA 編輯主要以胞嘧啶轉換成尿嘧啶的形式存在,且主要發生在密碼子的第一、二位堿基[32,35],但在二穗短柄草(Brachypodium distachyon)[36]中檢測到19個編輯位點發生在密碼子的第三位堿基,在小麥[37]中也存在1個編輯位點發生在第三位堿基上。自1991年,Hoch等[38]首次發現在玉米葉綠體蛋白編碼基因rpl2發生RNA編輯后,即蘇氨酸密碼子ACG轉變為起始密碼AUG,人們便對高等植物葉綠體的RNA編輯現象進行了大量研究。通過對小麥[37]、水稻、黑麥、甘蔗(Saccharum officinarum)、玉米、野生二粒小麥(Triticum dicoccoides)[39]、粗山羊草(Aegilops tauschii)[40]及大麥[41]等8種禾本科作物葉綠體的RNA編輯位點進行了比較分析,結果發現ndhA-158、ndhA-188、ndhB-156、ndhB-196、ndhB-204、ndhB-246、ndhB-277、ndhB-494、ndhD-295、rpoB-156、rpoB-182和rpoB-187等12個位點在8個物種中均發生了編輯;atpA-383、ndhA-357、rpl2-1、rps8-61和ycf3-62等5個位點在7個物種中發生編輯,表明這些位點容易發生編輯[39]。并且發現ndhB在這幾個葉綠體基因組中的編輯位點都最多[10,42]。研究發現RNA編輯的缺失可能會引起植物黃化,白化甚至幼苗致死[43-45]。例如,在擬南芥(Arabidopsis thaliana)中,RNA編輯位點rpoA-67和clpP-187的缺失導致植株黃化,幼苗致死[43];玉米綠色幼苗中ndhB第3個編輯位點發生編輯,而黃化幼苗中此位點不發生編輯[45]。
除此之外,通過對RNA編輯位點進行比較來分析物種間的進化關系,從而為禾本科物種的起源和進化研究奠定基礎。在ndhB-50、ndhB-235和ycf3-15 3個位點,二穗短柄草、黑麥和大麥都發生了編輯,而二穗短柄草和水稻只有在ndhB-235一個位點發生編輯,說明與水稻相比,二穗短柄草與黑麥和大麥的進化關系較近[36]。隨后,一些物種特異發生的編輯位點也被鑒定到,atpA-383只在野生二粒小麥和普通小麥中發生編輯;rpl2-1只在烏拉爾圖小麥(Triticum urartu)和野生二粒小麥中發生編輯;rps14-27只在普通小麥中發生編輯[46];atpF-47、atpF-127、atpB-1487、rpoA-386、rpoA-1009和rpoC2-2003的RNA編輯只發生在大麥中,表明葉綠體RNA編輯位點也存在一定的物種特異性[41]。
4 禾本科葉綠體基因組在系統發育組學中的應用
系統發育基因組學的研究是圍繞著系統發育學和基因組學展開的,主要研究內容包括在基因組水平上通過大量的分子數據研究生物之間的系統發育關系以及通過物種之間進化關系來進行基因組進化機制的研究[47-49]。由于線粒體基因組大小在各植物類群中變異很大,基因組中存在很多外源基因插入,且線粒體基因組中分子內重組的現象廣泛存在。所以目前,進行植物系統發育研究主要是利用葉綠體基因組和核基因組的基因組結構及變化進行分析。然而,在植物中由于核基因組的復雜性使得低拷貝基因的篩選比較困難。葉綠體基因組由于大小適中,基因組結構較為保守,易于測序。且各植物類群葉綠體基因組之間具有良好的共線性,便于比較分析。葉綠體基因組堿基替換率適中,近年來基于葉綠體基因組的系統發育基因組學得到了較快的發展,完整的葉綠體基因組序列對于破譯密切相關的類群之間的系統發育關系以及增進對植物物種進化的理解非常有價值[50]。
4.1 DNA條形碼在系統發育分析中的應用
DNA條形碼技術是利用生物體DNA中一個或幾個保守片段對物種進行快速準確鑒定的新興技術。葉綠體上的DNA序列片段(如matK、rbcL、trnH-psbA、rpoC1、rpoB、accD、ycf5和ndhJ等)在植物DNA條形碼被廣泛應用。Bieniek等[51]使用matK、rbcL和trnH-psbA 3個DNA條形碼對禾本科的小麥屬的系統發育進行研究,目的是評估由上述DNA條形碼標記(matK和rbcL)和候選標記(trnHpsbA)提供的系統發育信息的價值,并評估這些序列的物種識別效力。李永青等[52]通過對8種禾本科牧草DNA條形碼通用序列篩選得到matK(matK1、matK2和matK3)和rbcL基因的4個標記位點,并建立了相對應的特異DNA識別碼。研究可為混合禾本科牧草飼料中的高粱屬、玉蜀黍屬、芨芨草屬、針茅屬、黑麥草屬、羊茅屬和早熟禾屬的8種牧草準確識別提供分子水平上的科學依據。Song等[53]通過對4個高梁屬的葉綠體基因組之間的比較分析,得到651個可變位點,137個Indel和9個小倒位。并檢測到4個存在差異的DNA區域(rps16-trnQ、trnG-trnM、rbcL-psaI和rps15-ndhF),它們適合于系統發育和物種鑒定。系統發育分析得出高粱族是蜀黍族中的一個單族群。
4.2 葉綠體分子標記在系統發育分析中的應用
分子標記廣泛應用于遺傳育種、基因組作圖、基因定位、物種親緣關系鑒別、基因庫構建、基因克隆等方面的研究。目前,基于葉綠體DNA的分子標記主要有:非編碼區分子標記、cpSSR標記、SNP和cpInDel標記等。葉綠體基因組中的SSR對于解決緊密相關的類群之間的遺傳多樣性非常有用,因此,增加種間研究的能力,可與核基因組開發的SSR標記結合使用,以解決關系緊密的物種之間的系統發育關系[53]。基于測定的葉綠體基因組序列,Qiu等[54]為5種優質羊茅物種組裝了葉綠體基因組,并鑒定了結構變異和突變熱點,開發確定了cpSSR標記,以促進優良羊茅物種的鑒定。并結合羊草屬和黑麥草屬中其他物種的葉綠體基因組,重建了羊茅屬和黑麥草屬復合體的系統發育關系。
4.3 葉綠體基因組在系統發育分析中的應用
同種中,不同亞種的葉綠體基因組序列的不同,反映了它們在進化過程中產生了差異。葉綠體基因組數據庫的迅速充實為揭示葉綠體基因組進化理論的研究提供了更充足的條件。通過比較野生稻與栽培稻的葉綠體基因組序列,發現野生稻葉綠體全基因組中存在插入和缺失現象,多發生在 IR區域的編碼區,堿基的置換則發生在LSC和SSC區域[55]。Cheng等[56]對412份水稻種質包括野生稻、粳稻、秈稻的葉綠體基因組進行分析,揭示出亞洲水稻(秈稻和粳稻)與野生稻的分離簇,并結合中國[57]和印度[58]亞洲水稻的考古證據進行驗證,兩個結果都表明秈稻和粳稻在葉綠體基因組水平上可能具有獨特的背景,即亞洲水稻(Oryza sativa L.)至少被馴化了2次。
通過以煙草為對照,對玉米、水稻和小麥的106個葉綠體基因的核苷酸序列進行比較分析,表明玉米、水稻和小麥的葉綠體基因組的大多數基因區域進化速率相似,但RNA基因具有高度保守的進化速率,并通過葉綠體基因的可變核苷酸位點來構建系統發育樹,從而得出支持水稻和小麥之間的關系更加緊密的論點[59]。Givnish等[60]基于545個單子葉植物和22個亞群中的77個葉綠體基因組,對系統發生率進行了最大似然分析。研究表明物種多樣化經歷了4次大規模加速,禾本科(Poaceae)、多葉菊科(Danyanthaceae)、蘭亞科(Orchidoideae)的樹蘭亞科(Epidendroideae)和傘形科(Lemnoideae)的天南星科(Araceae),它們均與特定的生態/形態變化有關。單子葉植物的分支確定和支持隨著基因數目和分支長度的增加而增加,并隨著相對分支深度的增加而減少。
Mondal等[61]通過對短粒野生稻,對組裝的葉綠體基因組以及其他11個測序的稻屬物種的葉綠體序列進行了系統發育分析,推斷該新組裝的葉綠體基因組與稻屬中的粳稻亞組接近。Moner等[62]對58個野生和馴化的水稻樣品進行了完整的葉綠體基因組序列分析,以研究它們的系統發育,從而提供了有關全球主要野生A基因組水稻主要群體的生物地理學的更多信息。通過在AUS稻和粳稻進化枝中鑒定AUS(一種主要產于孟加拉國的水稻)種質,提出了栽培的AUS組水稻的多系母本基因組起源。當前葉綠體類型的分布似乎與核基因組多樣性的分布明顯不同,表明水稻祖先的復雜進化歷史導致了水稻的馴化。竹亞科作為禾本科的一個分支,由于其很少開花和無性繁殖的原因使得它不論從形態上還是分子水平上,都被認為是分類學中的一個困難群體。通過對24個完整的葉綠體基因組進行系統發育分析,為證實由竹亞科(Bambusoideae)與稻亞科(Ehrhartoideae)、早熟禾亞科(Pooideae)共同構成了BEP分支中竹亞科和早熟禾亞科為姊妹關系提供了有力支持[63]。
5 展望
白、疫苗、生物材料等,將為葉綠體基因組轉化技術的發展提供了廣闊的應用前景。
隨著基于高通量的第三代測序技術的不斷發展,禾本科作物葉綠體全基因組研究將不斷深入,測序將會更快速、更便宜,這有利于構建完整的植物DNA條形碼數據庫和公共序列數據庫,使DNA條形碼技術越來越實用[64]。不僅如此,新一代測序技術的發展,將促進葉綠體系統發育基因組學在較低級分類階元中的應用。這將極大推動葉綠體系統發育基因組學的發展,從而為植物的系統發育研究提供更多的條件支持。對葉綠體基因組的深入研究也將會為物種鑒定、轉基因、基因編輯及近緣物種的劃分等方面提供一定的理論依據。同時,也為禾本科作物的定向遺傳改良,創制新資源提供理論支撐。
葉綠體基因組結構相對穩定,含有大量遺傳信息,被視為探索植物間進化關系的重要數據來源。禾本科植物中含有較多的糧食作物和經濟作物,故其在葉綠體基因組學的研究較為密集。但禾本科植物在進化時葉綠體基因組中基因、內含子丟失的機制暫不明了;小麥、水稻、玉米等主要禾本科作物葉綠體基因組RNA編輯機制仍未明確;對已完成測序的物種,需要對其序列進行整合分析,對葉綠體基因組的結構和基因功能進行深入研究。
由于植物的遺傳信息更為復雜,物種間的雜交和進化可能會使不同物種帶有相同的cpDNA,同一物種也可能攜帶有不同的cpDNA,從而影響DNA條形碼的鑒定結果[64]。DNA條形碼技術應與生物學、細胞學和分子系統學相結合,在實際應用中,范圍維持在對某一科或某一屬的植物進行鑒定,才能減少誤差。運用葉綠體上的DNA序列片段所形成的DNA條形碼,不僅可以為作物近緣種屬親緣關系的判斷提供依據,也可以在作物育種中的品種或育種材料的衍生系鑒定等方面提供很大的幫助。在未來的育種工作中,一方面基于葉綠體基因組序列的DNA條形碼與基于核基因組序列的DNA條形碼相結合,用于品種保護、新品種選育和物種鑒定,這將大大降低成本,提高育種效率。另一方面,通過葉綠體遺傳轉化來提高作物產量,培育抗蟲、抗旱、抗鹽轉基因植物,以及利用植物葉綠體生產藥用蛋
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
青少年科技博覽(中學版)(2022年6期)2022-12-27 19:44:27
軍事文摘(2021年22期)2021-11-26 00:43:51
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
少兒科學周刊·兒童版(2015年10期)2015-11-07 03:42:03
