DNA堿基編輯器在微生物中的研究進展
2020-12-28 07:01:48劉振權董會娜叢麗娜張大偉
工業微生物 2020年6期
劉振權, 董會娜, 叢麗娜, 張大偉*
1.大連工業大學 生物工程學院,遼寧 大連 116034; 2.中國科學院天津工業生物技術研究所,天津 300308
CRISPR系統作為一種自身免疫系統在古細菌和細菌等原核生物中普遍被報道,該系統可對外源入侵的核酸序列靶向切割使其斷裂,從而起到保護機體免受入侵核酸序列干擾的作用[1]。Ⅱ型系統僅需一種Cas9效應蛋白即可發揮切割功能且具有較高活性。因此,CRISPR/Cas9作為基因編輯工具被廣泛研究與利用[2, 3],作為編輯工具的II型CRISPR/Cas9主要由以下兩個功能性原件組成,包括具有核酸酶活性的Cas9蛋白[4],具有靶向作用的sgRNA[5]。目前應用的CRISPR/Cas9系統主要來源于化膿鏈球菌[6]。Cas9的原間隔物相鄰基序(PAM)區一般位于目標位點的3'末端,序列是5′-NGG-3′[6, 7]。
CRISPR/Cas9工具具有高效性、簡便性、使用廣泛性等特點[8]。通過改變sgRNA的序列,實現對不同位點的切割[9, 10]。在進行基因編輯工作中,CRISPR/Cas9會導致雙鏈斷裂(DSB)的產生,因此需要修復系統進行修復[11, 12]。目前常用的修復系統有兩種,一種是容易出錯的不精確修復系統即非同源性末端連接(non-homologous end joining, NHEJ)[13],另一種是不易引起錯誤的精確修復系統即同源性末端連接(homology-directed repair, HDR)[14, 15]。利用NHEJ修復系統對CRISPR/Cas9系統產生的DSB進行修復時,會在DSB附近的基因序列中隨機引入非特異性的堿基缺失或者插入,從而導致非理性的編輯。利用CRISPR/Cas9與HDR系統構建的基因編輯工具,已經被成功地應用于眾多菌株中,實現了基因的精確編輯,包括堿基序列的插入、刪除以及單堿基的突變工作[16, 17]。但是該系統需要外源提供模板并且在一些編輯過程中可能留下篩選標記,同時由于HDR在一些生物中具有較低的效率,導致在一些生物中并不能發揮功能。在修復系統的效率不足以對CRISPR/Cas9引入的DSB進行修復時,CRISPR/Cas9的引入會導致生物死亡,導致無法現實編輯[18]。這也促進了CRISPR/dCas9系統的發展。
在CRISPR/Cas9系統中,通過突變切割結構域HNH(H840A)與RuvC(D10A),分別失活了對互補鏈與非互補鏈的切割功能,獲得不具有切割功能,僅具有靶向作用的CRISPR/dCas9系統[19]。利用CRISPR/dCas9介導的堿基編輯器獨立于宿主細胞自身的NHEJ和HDR系統,利用脫氨酶的催化活性實現堿基轉化[20-22]。基于CRISPR/dCas9系統與胞嘧啶脫氨酶(PmCDA1)融合的胞嘧啶脫氨酶系統(CBE),已在大腸桿菌中被成功建立,實現了由胞嘧啶核苷酸(C)到胸腺嘧啶核苷酸(T)的突變,突變率高達61.7%~95.1%,并且引入該系統不會導致細菌的死亡[14]。研究者通過單獨失活CRISPR/Cas9系統中一個切割結構域的功能,獲得CRISPR/nCas9系統。利用該系統融合脫氨酶構建堿基編輯器,在一定程度上提高了編輯效率[3]。關于腺嘌呤堿基編輯器的報道較少,GAUDELLI N M等人最先在大腸桿菌中對腺嘌呤脫氨酶進行定向進化,與CRISPR/dCas9融合表達構建了能夠實現腺嘌呤核苷酸(A)到鳥嘌呤核苷酸(G)突變的ABE編輯工具[23]。本文針將從原理、發展史、應用以及未來的優化方案對上述DNA堿基編輯器進行綜述。
1 堿基編輯器的原理
目前ABE和CBE是已經被開發的兩種DNA堿基編輯器。堿基編輯器由CRISPR/dCas9系統和脫氨酶系統組成。通過對Cas9基因突變(H840A、D10A),得到不具有切割功能,但仍然保留靶向作用的CRISPR/dCas9系統[24]。構成堿基編輯器的脫氨酶主要有兩種,一種是胞嘧啶脫氨酶,用于構建CBE工具,利用胞嘧啶脫氨酶的作用將胞嘧啶結構中的氨基轉化為氧原子,同時失去臨近氮原子上的氫原子,進而實現由C到尿嘧啶核苷酸(U)的變化。在DNA復制和修復過程中又將U轉化為T,進而完成C到T的轉換[24],另一種是腺嘌呤脫氨酶,用于構建ABE工具,利用腺嘌呤脫氨酶的作用將腺嘌呤核苷結構中的氨基轉化為氧原子,同時失去臨近氮原子上的氫原子,其將A轉換為次黃嘌呤核苷酸(I),在DNA復制和修復過程中又將I轉換為G,進而完成A到G的轉換。在上述兩種DNA堿基編輯器共同作用下可以實現四種堿基的轉換,即C到T;A到G;T到C以及G到A[23]。圖1 為DNA堿基編輯器的原理圖。
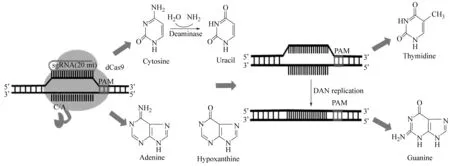
圖1 CBE和ABE原理圖
2 堿基編輯器的發展過程
2.1 CBE工具的發展過程
胞嘧啶堿基編輯器最早被應用在真核生物中,在原核生物中的開發利用報道較少。經過對dCas9與脫氨酶以及相關功能原件不斷的更新,目前已經開發到第四代堿基編輯器。其中第一代堿基編輯器由KOMOR A C等人[24]開發,研究者分別利用dCas9與小鼠來源的APOBEC1脫氨酶融合表達,實現了目標位點C到T的轉換。CBE1的可編輯框范圍是5 bp,編輯位點主要位于距離PAM遠端的13~17 bp處(距離PAM近端定義為第一位)。在生物體內其效率為0.8%~7.7%,顯然如此低的編輯效率,并不能滿足實驗需求。
為了提高編輯效率KOMOR A C等人繼續深入研究,開發了第二代堿基編輯器CBE2[24]。CBE2是在CBE1的基礎上融合表達了具有抑制DNA羰基化酶(Uracil N-glycosylase,UNG)活性的DNA羰基化酶抑制劑(Uracil Glycosylase Inhibitor,UGI),抑制了體內的修復系統提高了編輯效率。實驗結果顯示,CBE2在真核生物細胞中編輯效率仍然很低,但其在細菌中能夠有好的效果。
接下來,研究人員利用nCas9(D10A)代替了CBE1中的dCas9構建了第三代胞嘧啶堿基編輯器CBE3。nCas9可以形成一條鏈的斷裂,當靶向未被編輯的DNA單鏈時,nCas9復合物會造成未被編輯單鏈的斷裂,此時有機體必需利用另一條被編輯的鏈作為模板進行DNA修復。這個過程使得兩條鏈相應位置的堿基均發生了突變,以這兩條發生編輯的單鏈為模板進行DNA復制,則子代DNA均是發生編輯的。理論上會很大程度提高編輯效率。但是必需考慮到當引入nCas9后,有可能引起一部分細胞的死亡。實驗結果顯示,CBE3編輯器提高了真核生物的編輯效率,效率可達37%,但是同時發現該工具的引入會引起一些不想要的插入突變(Indel)[24]。
NISHIDA K等人,利用nCas9與七鰓鰻來源的胞嘧啶脫氨酶PmCDA1融合表達,構建了類似于CBE3的另一種堿基編輯器并命名為Target-AID[25]。由于不同來源的脫氨酶,導致該工具在一些特點上與CBE3具有一定區別。首先,Target-AID的可編輯范圍與CBE3有所不同,一般在PAM序列的遠端15-19 bp處。其編輯效率幾乎不受編輯位點臨近堿基種類的影響。Target-AID與CBE3都有C轉換為G的情況出現,但是與CBE3不同的是Target-AID工具將C突變為G是隨機的,并不像CBE3那樣與被編輯的C位置有關。
C突變為G并不是規律出現的,這種突變嚴重地影響堿基編輯工具的精準性。為了進一步提高堿基編輯器的精準性,KOMOR等人,通過對nCas9與脫氨酶之間linker以及nCas9與UGI之間linker長度的優化,同時過表達UGI,開發了第四代堿基編輯器CBE4。其與CBE3相比具有更高的編輯效率(是CBE3的編輯效率的1.5倍),以及更低的C到G的突變與Indel出現的頻率(與CBE3相比降低23倍)[26]。為了降低Indel的出現對基因表達造成的影響,KOMOR等人,在CBE4的基礎上,進一步表達了Gam蛋白,利用Gam蛋白對DNA缺口的保護作用進一步減少了Indel的出現頻率。
KIM Y B等人,在CBE3的基礎上對APOBEC1進行點突變,得到的突變體有效地縮小了編輯框的范圍,由原來的5 bp減少到現在的1 bp~2 bp。在一定程度上提高了堿基編輯器的精準性,尤其是當編輯位置存在多個胞嘧啶的時候,會顯著地提高編輯的精準性。PAM的種類很大程度地限制了堿基編輯器的使用范圍。對于一些不具有合適PAM序列的位點,CBE3是無法發揮作用的。因此KIM Y B等人,對nCas9(D10A)進行突變,得到了可以識別不同PAM序列的突變體。對之前一些無法作用的位點實現了編輯,一定程度上提高了CBE工具應用的廣泛性[27]。后續相關研究者針對于PAM進行了改進,用以增加CBE工具的可識別范圍,通過突變等手段一定程度上拓寬了CBE的編輯范圍[28-32]。
2.2 ABE工具的發展過程
ABE編輯工具由GAUDELLI等開發,他們認為如果腺嘌呤核苷脫氨酶代替CBE中的胞嘧啶脫氨酶,極大可能開發出ABE編輯工具。因此YANG L等人,嘗試目前已有的脫氨酶,但是均不能發揮作用[33]。對大腸桿菌來源的腺嘌呤脫氨酶TadA進行人工改造,獲得可以發揮功能的腺嘌呤脫氨酶TadA*,利用nCas9的靶向作用,首次成功地構建了ABE編輯工具。逐步地對ABE編輯工具中的脫氨酶進行突變,提高了ABE編輯工具的編輯效率。研究者在此基礎上對ABE進行了改進提高了編輯效率并增加了PAM可識別區域[34]。
GAUDELLI N M等人對ABE7.10進一步優化獲得了ABE8,與ABE7.10相比具有更高的編輯效率,更低的脫靶率[35]。最近RALLAPALLI K L等人利用計算機分子動力學模擬實驗揭示了在DNA編輯中TadA*的結構和功能。分析證實了該單一突變在賦予TadA*功能方面的重要性,并證明TadA*作為單體而非二聚體進行DNA堿基編輯[36]。ABE編輯工具與CBE編輯工具有所不同,CBE編輯工具中,脫氨酶在dCas9的下游融合表達。但ABE編輯工具脫氨酶位于dCas9下游表達時,ABE編輯工具無法發揮功能。當將dCas9基因在脫氨酶基因下游融合表達時,可以發揮A到G的編輯功能[23]。
3 堿基編輯器在微生物中的應用
3.1 利用CBE工具實現單位點多位點編輯工作
CRISPR/Cas9系統具有高效性,簡便性等特點,在真核生物與原核生物中得到廣泛應用。但是該系統在一些微生物中會導致生物體死亡,無法發揮編輯作用[18]。利用不具有切割作用的CRISPR/dCas9與胞嘧啶脫氨酶共同作用,在不影響微生物生長的情況下,現實了基因的編輯工作[37]。BANNO S等人利用脫氨酶融合核酸酶缺陷的CRISPR/Cas9系統的CBE編輯工具首次在大腸桿菌中靶向地實現了C突變T的研究。并證明了該工具的可編輯范圍是距離PAM序列遠端的15 bp~19 bp,并且編輯范圍受sgRNA長度的影響。通過添加尿嘧啶DNA糖基化酶抑制劑與降解標簽(LVA標簽)提高了該工具的編輯效率。并證明了該工具可以實現基因組的多位點編輯,同時實現了對大腸桿菌中六個不同的結構基因進行編輯[14]。
同年,WANG Y等人利用CRISPR/dCas9和胞嘧啶脫氨酶融合表達,在谷氨酸棒狀桿菌中利用尿嘧啶磷酸核糖轉移酶upp基因作為編輯靶點,證明了CBE編輯工具可以發揮編輯作用。在此基礎上深入研究實現了多位點的編輯工作,并證明了單位點、雙位點和三位點編輯效率分別高達100%、87.2%和23.3%[38]。同年,研究者證明了CBE編輯系統可實現銅綠假單胞菌,銅綠假單胞菌,惡臭假單胞菌和熒光假單胞菌等高效假單胞菌物種的基因失活和點突變[39]。
3.2 利用CBE工具實現基因失活工作
研究者選擇大腸桿菌菌株XL1-Blue作為模型,通過將CAG/CAA(Gln)或CGA(Arg)密碼子將轉換為各自的TAG/TAA/TGA終止密碼子,提前終止了翻譯過程,實現了對四環素抗性基因tetA的失活。通過流式細胞儀和X-gal細胞化學分析,顯示了99.93%被編輯過的大腸桿菌細胞失去熒光,表明BE3介導的大腸桿菌基因編輯效率幾乎為100%。與Cas9相比BE3蛋白在靶向和切割基因組時具有非致命性。同時證明了CBE編輯工具在苜蓿芽孢桿菌中,可靶向實現編碼氨基酸的密碼子突變為終止密碼子,從而廢除了蛋白質功能[37]。使用設計的sgRNA將谷氨酸棒狀桿菌的尿嘧啶磷酸核糖轉移酶upp基因第484位的C轉換為T生成終止密碼子,并利用其突變體證明了失活該基因的編輯效率高達11.2%[38]。
肺炎克雷伯菌是一種有潛力的工業微生物,也是人類的主要病原體。研究者在肺炎克雷伯菌中利用CBE編輯工具,在不產生DSB,不需要修復模板的情況下,通過將四個密碼子(CAA,CAG,CGA和TGG)轉化為終止密碼子而實現了基因失活。并證明了基因距離PAM的位置對于編輯效率的影響,距離PAM序列遠端的12 bp~17 bp的TC基序的C轉化為T的效率幾乎為100%,而其他位置C的編輯效率則低。同時最靠近胞嘧啶上游的堿基種類對編輯效率也有影響。胞嘧啶上游是胸腺嘧啶T時編輯效率最高C而A次之,上游是G時編輯效率最低。TC的編輯效率高于CC和AC的編輯效率。GC的編輯效率最低[40]。
由于耐藥性金黃色葡萄球菌的出現,迫切需要開發針對金黃色葡萄球菌感染的新型治療手段。研究者設計了一個Cas9切口酶(Cas9D10A)和一個APOBEC1的融合體,利用CBE編輯工具實現高效的基因失活和金黃色葡萄球菌的點突變。利用該工具將agrA基因第7位的C,cntA基因第5位的C和esaD基因第6位的C成功以100%的效率突變為T,產生終止密碼子從而提前終止了翻譯過程。并證明該工具可編輯的范圍是距離PAM序列遠端的12 bp~16 bp。相鄰堿基對編輯器的編輯效率有一定影響,體外活性實驗效率由高至低遵循TC、CC、AC和GC的規律[41]。
3.3 ABE工具的應用
ABE工具已經在人類和動植物中發揮作用,實現了由A/T到G/C的突變,但是相關研究較少,且效率沒有CBE編輯工具高[42-44]。在微生物中報道的極少,目前僅GAUDELLI N M等人在大腸桿菌中對腺嘌呤脫氨酶進行定向進化和蛋白質工程產生了第七代ABE,首次證明了腺嘌呤堿基編輯器可以在微生物中發揮功能,將目標A/T堿基對有效地轉化為G/C[23]。研究者在小鼠中建立ABE編輯工具時,為優化ABE編輯工具的編輯效率與可作用范圍,分別利用金黃色葡萄球菌 (Staphylococcusaureus)、釀膿鏈球菌(Streptococcuspyogenes)來源的腺嘌呤脫氨酶進行實驗比傳統的ABE編輯工具效率更高,可作用范圍更廣,這也為在細菌中建立ABE編輯工具提供了希望[45]。
4 展望
由于堿基編輯器的簡便性,高效性,不需要額外的修復系統且不導致菌體死亡等特點已被廣泛應用與真核與原核生物中,但是該工具仍有一些不足之處。譬如,在編輯過程中的脫靶率較高;可以編輯的范圍會受到一定限制例如PAM的限制;在靶向位點臨近處有相同堿基時,很難實現某一個堿基的突變。這些問題的解決仍然是堿基編輯器進一步優化的方向。在堿基編輯器中主要是CRISPR系統發揮靶向作用,所以在解決堿基編輯器脫靶率的問題上,可以參考CRISPR系統脫靶率問題的解決方法。可以利用生物信息學的方法,對sgRNA進行設計與篩選[46]。通過改變sgRNA的長度在一定程度上降低了脫靶率[47]。通過對dCas9蛋白的結構域進行突變獲得具有識別不同PAM的dCas9蛋白[27, 47],可以在一些特異位點利用ABE工具實現C到A/T的轉換[48]。同時,可以利用CRISPR/Cas12a與脫氨酶融合構建堿基編輯器[49,50]。從目前的報道中看,可編輯的范圍只在靶向序列范圍內,可以適當縮短靶向序列的長度,一定程度上縮小了可編輯的范圍,進而實現了單個堿基的突變[47]。
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
裝備制造技術(2019年12期)2019-12-25 03:06:46
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
家庭影院技術(2017年9期)2017-09-26 03:41:45
商周刊(2017年9期)2017-08-22 02:57:49
時代英語·高二(2015年1期)2015-03-16 00:08:11
