NF-κB-iNOS/COX-2信號通路在高糖損傷血管內皮依賴性舒張中的作用*
2021-01-06 08:26:44邱紅梅蔣青松
中國病理生理雜志 2020年12期
關鍵詞:糖尿病
陽 創,薛 萊,吳 陽,邱紅梅,蔣青松△
(1重慶醫科大學藥學院藥理教研室,重慶市生物化學與分子藥理學重點實驗室,重慶 400016;2江油市人民醫院臨床醫學部,四川江油 621700;3中山大學附屬第七醫院心血管中心,廣東深圳 518107)
糖尿病是全球高發的慢性疾病之一。2019年全球糖尿病患者已達4 億多人;據國際糖尿病聯盟估計,至2045 年,這個數字會上升到7 億[1]。血管病變是糖尿病及其并發癥發生發展的關鍵因素,也是導致糖尿病患者死亡的首要因素之一[2]。糖尿病血管并發癥包括微血管并發癥如糖尿病視網膜病、糖尿病心肌病和糖尿病腎病等,大血管并發癥有中風、冠心病和外周血管疾病等[3]。糖尿病血管病變的參與因素眾多,血管內皮結構損傷從而導致內皮功能障礙是其關鍵的病理生理基礎[4]。
乙酰膽堿(acetylcholine,ACh)引起的內皮依賴性血管舒張是檢測血管內皮功能的重要指標,其功能障礙是血管內皮損傷的初始表現之一[5]。現有研究認為,長期全身慢性低水平炎癥是促進糖尿病發生發展的重要因素。通過不同信號通路,炎癥反應促進內皮細胞損傷,引起內皮功能障礙[6]。核因子κB(nuclear factor-κB,NF-κB)是細胞內重要的核轉錄因子,參與機體的炎癥反應、免疫應答及腫瘤形成等多種過程。NF-κB 調控與炎癥有關的基因,在糖尿病及其并發癥中有重要作用[7]。內皮細胞分泌多種血管活性物質以維持正常的血管功能,包括舒血管物質如一氧化氮(nitric oxide,NO)、前列環素(prostacyclin,PGI2)和內皮衍生超極化因子等,及縮血管物質如內皮素-1、血管緊張素Ⅱ和血栓烷A2(thromboxane A2,TXA2)等[8]。其中,NO 和PGI2/TXA2是最為重要的調控因素之一。誘導型一氧化氮合酶(inducible nitric oxide synthase,iNOS)和環氧合酶2(cyclooxygenase,COX-2)是NF-κB 的重要下游因子,也是NO 水平和PGI2/TXA2平衡調節的關鍵酶。目前對NF-κB-iNOS/COX-2 信號通路的研究主要集中在炎癥和腫瘤等方面[9],對于其在糖尿病內皮損傷的研究較少,對高糖條件下內皮依賴性血管舒張中的作用目前國內外未見報道。
本研究擬采用高糖孵育大鼠胸主動脈環建立糖尿病血管內皮損傷模型[10],分別給予NF-κB、iNOS和COX-2 抑制劑,觀察ACh 誘導內皮依賴性血管舒張作用的變化,以探討NF-κB-iNOS/COX-2 信號通路在糖尿病內皮損傷早期的作用。
材料和方法
1 動物
SD 大鼠,SPF 級,體重250~260 g,雌雄各半,由重慶醫科大學實驗動物中心提供,許可證號為SYXK(渝)2012-0001。
2 主要試劑
苯腎上腺素(phenylephrine,PE)購自上海禾豐制藥有限公司(批號H31021175);ACh、二硫代氨基甲酸吡咯烷(pyrrolidine dithiocarbamate,PDTC)、S-甲基異硫脲硫酸鹽(S-methylisothiourea sulfate,SMT)和BCA 蛋白濃度測定試劑盒均購自江蘇省碧云天生物技術研究所;美洛昔康(meloxicam)購自大連美侖生物技術有限公司;Trizol、逆轉錄試劑盒、SYBR Green Supermix 和PCR 引物購自TaKaRa;兔抗大鼠phospho-NF-κB p65 單克隆抗體購自Cell Signal?ing Technology;兔抗大鼠iNOS 多克隆抗體購自Ab?cam;兔抗大鼠COX-2 多克隆抗體購自Proteintech;兔抗大鼠β-actin多克隆抗體購自博奧森生物科技公司;ECL 化學發光試劑盒購自Thermo。其余試劑為國產分析純。
3 方法
3.1 內皮依賴性血管舒張功能的檢測[10]將SD 大鼠脫頸處死后立即取出胸主動脈,制成長3~4 mm的血管環,置于含20 mL Krebs 液[成分(mmol/L):NaCl 119,CaCl22.5,KCl 4.7,MgSO41.2,KH2PO41.2,NaHCO325,glucose 11,pH 7.4]的浴槽中37℃保溫,持續通以0.95 O2+0.05 CO2混合氣體。血管張力經張力傳感器(5 g,中國北京航天醫學工程研究所)連接于BL-420S 生物信號處理系統(成都泰盟科技有限公司),靜息負荷1 g。每隔15 min 更換Krebs液1次,平衡60 min后加入PE 1 μmol/L 預收縮血管,達到坪值并穩定后,加入ACh 30 μmol/L 驗證內皮的完整性(舒張率大于60%認為內皮功能完好)。換液,平衡30 min 后,再次用PE 預收縮血管。達到相似收縮坪值后,移除PE。然后將血管環置于不同條件(見實驗分組)的Krebs液中孵育6 h后,Krebs液清洗移去藥物,再次給予PE 收縮血管,達到孵育前相似收縮坪值并穩定后,按濃度梯度法給予ACh(0.5對數單位遞增,0.001~30 μmol/L),觀察血管環的舒張作用,計算血管最大舒張效應(Emax,血管最大舒張作用與PE 收縮力的百分比)和pD2(ACh 產生50%Emax濃度的負對數)。
實驗共分為5 組:正常糖(normal glucose,NG)組:葡萄糖11 mmol/L,加入甘露醇44 mmol/L 調節滲透壓;高糖(high glucose,HG)組:葡萄糖55 mmol/L;NF-κB 抑制劑PDTC(1 μmol/L)組、iNOS 抑制劑SMT(10 μmol/L)組和COX-2 抑制劑美洛昔康(100 μmol/L)組:高糖培養并加入相應抑制劑。每組重復6次。
3.2 HE 染色和電鏡觀察 將不同條件孵育血管環4 %多聚甲醛固定24 h,梯度濃度乙醇和二甲苯脫水,石蠟包埋,作3~5 μm冠狀切片,HE染色,于高倍鏡下觀察血管組織形態學變化。
或戊二醛固定2 h,磷酸液漂洗,鋨酸液固定后,用不同濃度乙醇和丙酮脫水、包埋,烘箱過夜。用超薄切片機作50~60 nm 切片,3%醋酸鈾-枸櫞酸鉛雙染色,透射電鏡下觀察血管超微結構變化。
3.3 RT-qPCR 檢測mRNA 的表達 取液氮凍存血管組織,按照RNAiso Plus Total RNA 提取試劑說明書的操作程序提取總RNA。參照GenBank 中大鼠基因序列,由重慶醫科大學生物化學與分子藥理學重點實驗室設計引物,經TaKaRa 生物技術公司合成并驗證(引物序列見表1)。按照TaKaRa Prime ScriptTMRT Reagent Kit with gDNA Eraser 試劑盒說明書操作,逆轉錄合成cDNA。反應條件為:95℃30 s;95℃5 s,60℃30 s,40 個循環。以β-actin 為內參照,對熒光強度達到閾值時的循環數(即Ct值)進行統計。根據相對定量公式進行計算。每組重復3次。
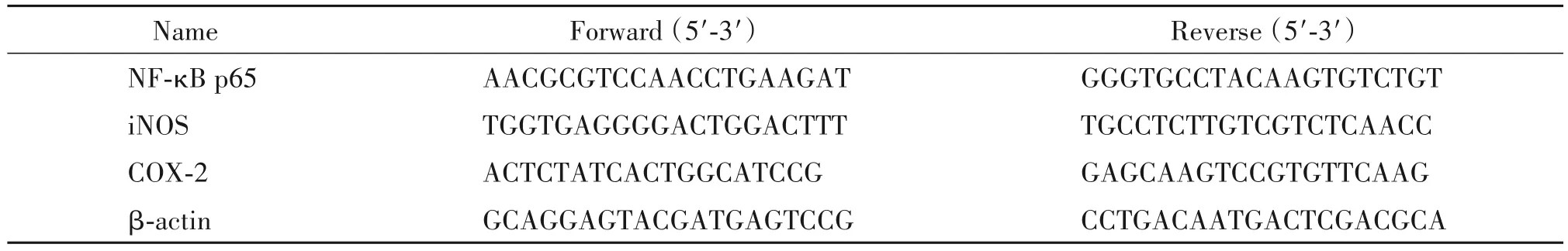
表1 RT-qPCR引物序列Table 1.The sequences of the primers for RT-qPCR
3.4 Western blot 檢測蛋白表達 血管組織液氮冷凍后勻漿提取總蛋白,BCA 法檢測蛋白濃度。取30 μg 蛋白進行SDS-PAGE 分離,轉膜,封閉,加入Ⅰ抗[β-actin(1∶5 000)、phospho-NF-κB p65(1∶1 000)、COX-2(1∶500)和iNOS(1∶500)],4℃過夜;洗膜,加入辣根過氧化物酶標記的Ⅱ抗(1∶3 000),洗膜后用ECL 發光試劑顯影,用Image Lab 軟件進行圖像分析。以目標蛋白與β-actin條帶的灰度比值表示蛋白表達的相對水平。每組重復3次。
4 統計學處理
采用SPSS 20.0 軟件進行統計學分析,用Excel和GraphPad Prism 5.01 等軟件作圖。實驗數據以均數±標準差(mean±SD)表示。多組間均數比較采用單因素方差分析(one-way ANOVA),組間兩兩比較采用SNK-q檢驗。以P<0.05為差異有統計學意義。
結 果
1 高糖對大鼠胸主動脈組織形態學的損傷及PDTC的作用
HE 染色和電鏡觀察結果顯示,正常組的血管結構清晰完整,血管平滑肌細胞排列整齊,內皮細胞完整平坦,內外彈力層連接緊密;高糖組孵育6 h后,血管結構破壞,內皮細胞受損、腫脹,內彈性膜斷裂,內層平滑肌細胞排列疏松紊亂;給予PDTC(1 μmol/L)后,血管形態顯著改善,平滑肌細胞排列較整齊,內皮細胞損傷減輕,見圖1。
2 不同抑制劑對高糖損傷ACh 血管內皮依賴性舒張作用的影響
高糖孵育6 h 后,HG 組血管環的PE 收縮力與NG 組相比差異無統計學顯著性(P>0.05);但在高糖環境下,ACh誘導的血管內皮依賴性舒張明顯減弱,其Emax和pD2均顯著降低(P<0.01),見圖2、表2。
不同抑制劑與高糖共孵育后,PE 的收縮力亦無明顯變化(P>0.05);但PDTC(1 μmol/L)、SMT(10 μmol/L)和美洛昔康(100 μmol/L)均明顯改善高糖損傷的血管內皮依賴性舒張功能,其Emax和pD2均顯著升高(P<0.01),見圖3、表2。
3 PDTC 對高糖條件下大鼠胸主動脈NF-κB p65、iNOS和COX-2 mRNA和蛋白表達的影響
與NG 組比較,高糖孵育后NF-κB p65、iNOS 和COX-2 的mRNA 和蛋白水平均顯著上調(P<0.01);給予PDTC(1 μmol/L)后,與HG 組比較,NF-κB p65、iNOS和COX-2的mRNA和蛋白表達水平則顯著下降(P<0.01),見圖4、5。
討 論
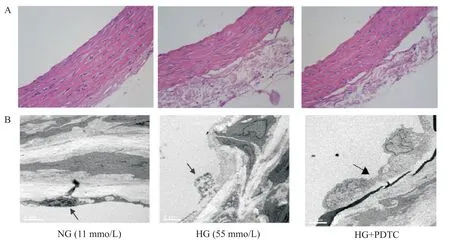
Figure 1.The histopathological changes of rat aortic ring.A:HE staining(×400);B:electron microscope(×7 000).The black ar?rows indicated endothelium.圖1 大鼠胸主動脈組織的病理變化
由于胰島素分泌絕對/相對不足,高糖血癥是糖尿病患者的主要特征,常伴特異性大血管和微血管并發癥。血管病變在糖尿病及其并發癥的發生發展中具有重要意義。研究已經表明,持續高糖狀態會損傷血管內皮,出現血管內皮功能障礙[11]。本研究中,與相同滲透壓的正常濃度(11 mmol/L)葡萄糖組相比,高濃度(55 mmol/L)葡萄糖使ACh 誘導的血管內皮依賴性舒張功能明顯下降至(16.73±6.81)%水平,遠低于內皮完整的60%的標準,提示血管內皮功能嚴重受損,且該損傷作用不是由于滲透壓增加引起。同時,病理觀察也證實高糖破壞了血管內皮的正常結構;另外,高糖對PE 所誘導的血管收縮沒有明顯影響,提示本實驗條件下,高糖可能未損傷血管的收縮功能。大量研究認為,內皮功能紊亂和缺失是糖尿病血管病變的主要危險因素。因此,深入研究高糖條件下血管內皮損傷的參與因素和相關信號通路的作用,對于糖尿病及其并發癥的防治具有重要意義。
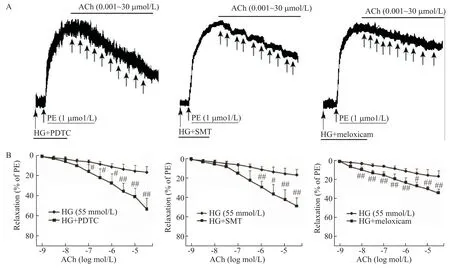
Figure 3.The effects of different inhibitors on the impaired endothelium-dependent relaxation induced by acetylcholine(ACh)under high glucose(HG)condition.A:the typical traces of the dose-response relationship for ACh-induced relaxation of rat aor?ta;B:the dose-response curves of ACh on aortic rings.Mean±SD. n=6.#P<0.05,##P<0.01 vs HG group.圖3 不同抑制劑對高糖損傷乙酰膽堿內皮依賴性舒張血管作用的影響
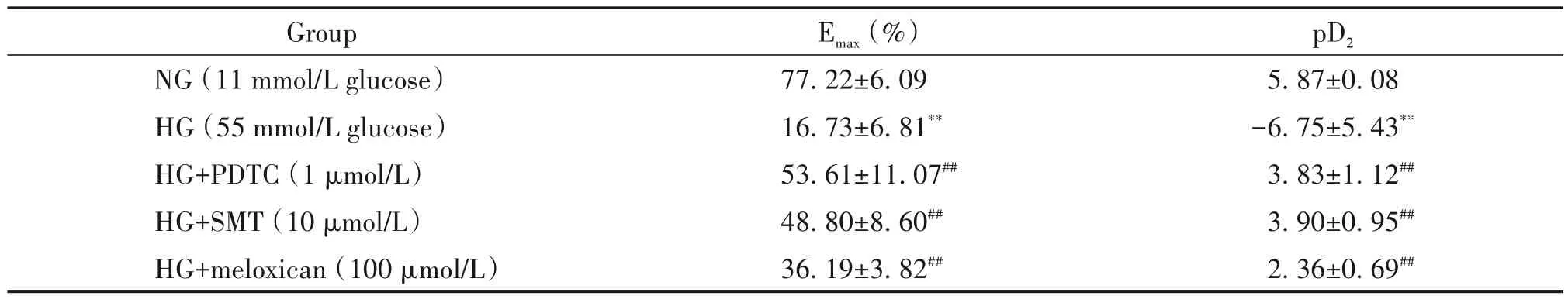
表2 不同抑制劑對高糖損傷內皮依賴性血管舒張的影響Table 2.The effects of different inhibitors on Emax and pD2 of acetylcholine-induced endothelium-dependent relaxation under high glu?cose(HG)condition(Mean±SD. n=6)
現有研究發現,炎癥是引起血管壁改變,導致內皮功能障礙的重要因素。糖尿病時,持續高血糖狀態使晚期糖基化終末產物(advanced glycation endproducts,AGEs)增加,AGEs 與內皮細胞和平滑肌細胞上的RAGE 受體結合,激活NF-κB,上調腫瘤壞死因子α(tumor necrosis factor-α,TNF-α)和白細胞介素1β(interleukin-1β,IL-1β)等多種細胞因子的表達,導致血管內皮細胞發生凋亡和炎癥,引發血管疾病[12-13]。NF-κB 是多條炎癥通路的交匯點,與多種炎癥相關紊亂的發生關系密切。生理情況下,NF-κB以無活性形式存在于胞漿中。病理狀態下,NF-κB迅速活化并移位至胞核,啟動一系列炎癥相關基因的轉錄。NF-κB p65 蛋白是NF-κB 的關鍵轉錄亞基之一,p65/p50異源二聚體是NF-κB最常見的活化形式。Kassan 等[14]的研究發現,NF-κB 活化損傷了糖尿病小鼠的血管功能,但NF-κB 對高糖作用內皮依賴性舒張影響的研究較少。本實驗結果顯示,高糖預孵育后,血管組織NF-κB p65的mRNA和蛋白表達水平均顯著升高,而NF-κB 抑制劑PDTC 下調其mRNA 和蛋白表達同時,也明顯恢復ACh 誘導的內皮依賴性舒張功能,改善高糖損傷的血管結構。這些結果提示,對于糖尿病初期內皮功能障礙的發生,NF-κB的活化可能也有重要作用。
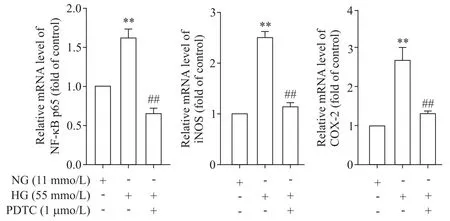
Figure 4.The effect of PDTC on the mRNA expression of NF-κB p65,iNOS and COX-2 in rat thoracic aorta under high glucose(HG)condition.Mean±SD.n=3.**P<0.01 vs NG group;##P<0.01 vs HG group.圖4 PDTC對高糖作用下大鼠胸主動脈NF-κB p65、iNOS及COX-2 mRNA表達的影響
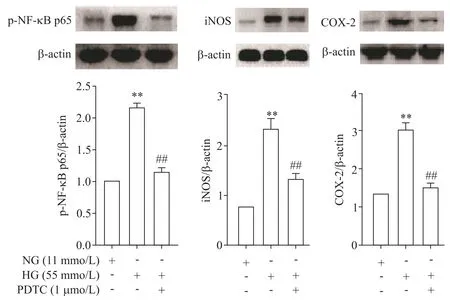
Figure 5.The effect of PDTC on the protein levels of p-NF-κB p65,iNOS and COX-2 in the rat thoracic aorta under high glucose(HG)condition.Mean±SD.n=3.**P<0.01 vs NG group;##P<0.01 vs HG group.圖5 PDTC對高糖作用下大鼠胸主動脈p-NF-κB p65、iNOS及COX-2蛋白水平的影響
NF-κB 不僅調節重要炎癥細胞因子的表達,也干預炎癥反應相關因子的合成,如iNOS 和COX-2[15]。正常情況下,iNOS 在血管內皮細胞低水平表達。但在炎癥等病理狀態時,iNOS 表達水平明顯增高,誘導內皮細胞持續生成大量NO。過量的NO 與超氧陰離子(·O2?)結合,生成過氧亞硝基陰離子,引起氧化應激反應,損傷血管內皮[16]。與iNOS 相似,生理狀態下,COX-2在多數組織不表達,但受到細胞因子、感染等因素誘導時,表達上調。當血管內皮細胞COX-2 表達上調,PGI2合成酶活性下降;同時,TXA2釋放增加[17]。PGI2/TXA2二者平衡失調是導致內皮依賴性舒張功能下降的重要因素。雖然已經證實,iNOS和COX-2是NF-κB重要的下游效應因子,但對于三者在高糖作用內皮依賴性舒張中的關系目前國內外未見報道。本研究中,高糖使內皮依賴性舒張功能顯著降低同時,iNOS 及COX-2 的mRNA 和蛋白表達水平均顯著上升;NF-κB抑制劑PDTC改善內皮結構和功能,并下調iNOS 及COX-2 的mRNA 和蛋白表達,提示iNOS 和COX-2 可能也介導了高糖激活NF-κB 損傷血管內皮的作用。值得注意的是,選擇性iNOS 抑制劑SMT 和COX-2 抑制劑美洛昔康均只能部分恢復ACh 誘導的內皮依賴性舒張作用,提示iNOS 和COX-2 相關信號通路作用共同參與高糖損傷的內皮依賴性舒張功能。另外,PDTC 也不能完全恢復高糖損傷的內皮依賴性舒張,提示除了NFκB-iNOS/COX2 通路之外,可能還有其他機制參與高糖血癥血管內皮依賴性舒張的調控。
綜上所述,本研究結果提示NF-κB-iNOS/COX-2通路參與了高糖損傷血管內皮依賴性舒張的作用。該通路的激活在糖尿病內皮損傷早期可能有關鍵作用,對該通路各靶點的干預對于糖尿病血管病變的防治可能有重要意義。但由于糖尿病血管病變發生參與因素的復雜性,高糖損傷血管內皮依賴性舒張的調控機制仍然需要更進一步的研究。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年3期)2021-08-22 06:49:56
中老年保健(2021年11期)2021-08-22 03:15:16
中國生殖健康(2020年2期)2021-01-18 02:51:44
中國生殖健康(2018年2期)2018-11-06 07:11:04
基層中醫藥(2018年2期)2018-05-31 08:45:04
