lncRNASNHG14/miR-331-3p/CCR1 軸在急性肺損傷中的作用機制研究
2021-01-12 06:19:36朱金源白吉佳張小亞
世界最新醫學信息文摘 2020年97期
關鍵詞:小鼠
朱金源,白吉佳,張小亞
(寧夏醫科大學總醫院重癥醫學科,寧夏 銀川)
0 引言
急性肺損傷及其最嚴重的階段急性呼吸窘迫綜合征,是嚴重威脅患者生命的常見臨床危重癥,發病機制復雜,迄今尚未完全闡明,且病死率仍高達40%[1]。因此,探索急性肺損傷的發病機制及有效治療措施頗有必要。LncRNA是一類新興的大分子非編碼 RNA,它與 miRNA 及其下游靶基因之間的相互調控作用同炎性疾病的發生密切相關。miRNA 作為一個轉錄后調控的重要因子,其活性可被lncRNA 通過“海綿”吸附的方式調控,此類 lncRNA 又被稱為競爭性內源 RNA。lncRNA 作為競爭性內源 RNA 競爭性地與 miRNA 結合,從而調節編碼基因的蛋白質水平,并參與調控細胞的生物學行為。lncRNA 可作為競爭性內源RNA 吸附 miRNA 調控肺上皮細胞凋亡,然而在急性肺損傷中發揮競爭性內源功能的 lncRNA 目前仍知之甚少,因此,我們希望能進一步探究競爭性內源機制在急性肺損傷中的作用。
lncRNA SNHG14 在脂多糖誘導的小鼠肺泡巨噬細胞中高表達,且作為競爭性內源RNA 與miR-34c-3p 和WISP1具有靶向調控關系,下調SNHG14 可通過miR-34c-3p 介導WISP1,從而緩解脂多糖誘導的急性肺損傷,說明SNHG14/miR-34c-3p/WISP1 軸在急性肺損傷的發生發展中起到一定的作用[2]。我們預實驗結果表明:脂多糖誘導后小鼠肺泡巨噬細胞和小鼠肺組織中miR-331-3p 表達降低,說明miR-331-3p 可能也參與了急性肺損傷的進程,但具體機制尚不明確。因此,我們猜測SNHG14 可能通過作為競爭性內源吸附miR-331-3p,進而促進急性肺損傷的發生。
1 資料與方法
1.1 材料
1.1.1 實驗動物
健 康 雄 性SPF 級C57/BL6 小 鼠54 只,6~8 周 齡,體 質量(20±2)g,購于南京模式動物所。實驗開始前7d 飼養于寧夏醫科大學總醫院動物實驗室,室溫(24±1)℃,相對濕度40%~80%,給予正常飲食,自由飲水。
1.1.2 主要試劑和儀器
MH-S 細胞購自美國ATCC 公司,LPS 購自上海勁馬生物科技有限公司,LightCycler 480II 實時熒光定量PCR 儀(瑞典Roche 公司)。
1.2 方法
1.2.1 miR-331-3p 的篩選以及與CCR1 靶向關系研究
(1)體外實驗ALI 細胞模型的建立和細胞轉染
體外培養小鼠肺泡巨噬細胞(MH-S),按照常規方法進行細胞培養換液,調整細胞濃度為4×105 個/mL,每孔100μL接種于96 孔培養板中培養1h。對照組加入12K 培養基,除空白對照組外其余各組加入終濃度為100μg/mL 的LPS,培養18h,構建ALI 細胞模型。根據細胞生長速度調整密度并鋪6 孔板,使細胞在第二日轉染時密度達80%-90%,使用lipofectmine2000(Invitrogen)試劑盒進行轉染。
(2)熒光定量PCR 方法檢測急性肺損傷細胞模型中miR-331-3p 的表達水平
通過生物信息學篩選CCR1 上游miR-331-3p,檢測ALI細胞模型中miR-331-3p 的表達水平。按Trizol 方法說明書操作提取總RNA,設計miR-331-3p 和CCR1 引物,由Takara公司合成。使用PrimeScriptRT 試劑盒將RNA 逆轉錄成cDNA,設定反應條件為:37℃,15min×3 次(逆轉錄反應),85℃5s(逆轉錄酶失活反應)。取反應液進行熒光定量PCR,參照SYBR?PremixExTaqTMII 試劑盒(RR820A,TaKaRa)說明書進行PCR 操作。
(3)Elisa 檢測各組炎癥因子的表達水平
調整細胞濃度為4×105 個/mL,每孔500μL 接種于24孔培養板中培養1h,加入終濃度為100μg/mL 的LPS,繼續培養24h。收集各孔細胞培養液于離心管中,1500r/min 離心10min,取上清液,按照細胞因子ELISA 檢測試劑盒的程序操作,檢測CRP、PCT、IL-1、IL-6、IL-8 和TNF-α 的表達。
1.2.2 SNHG14 吸附miR-331-3p 調控CCR1 的表達,參與ALI 進程
使用生物信息學軟件預測SNHG14 與miR-331-3p 的結合位點,采用雙熒光素酶試劑盒驗證SNHG14 與miR-331-3p 的靶向關系。用Promega 公司的Dual-Luciferase Reporter Assay System 檢測熒光強度。
1.2.3 體內實驗驗證SNHG14 調控CCR1 的表達,參與ALI 進程
SPF 級雌性C57BL/6 小鼠54 只,8~10 周齡,隨機分為Model 組( 鼻 內 滴 注LPS,1mg 溶 于5mL,N=120) 和Control組(鼻內滴注等體積生理鹽水,N=15)。qRT-PCR 檢測各組SNHG14、miR-331-3p 和CCR1 的表達情況。
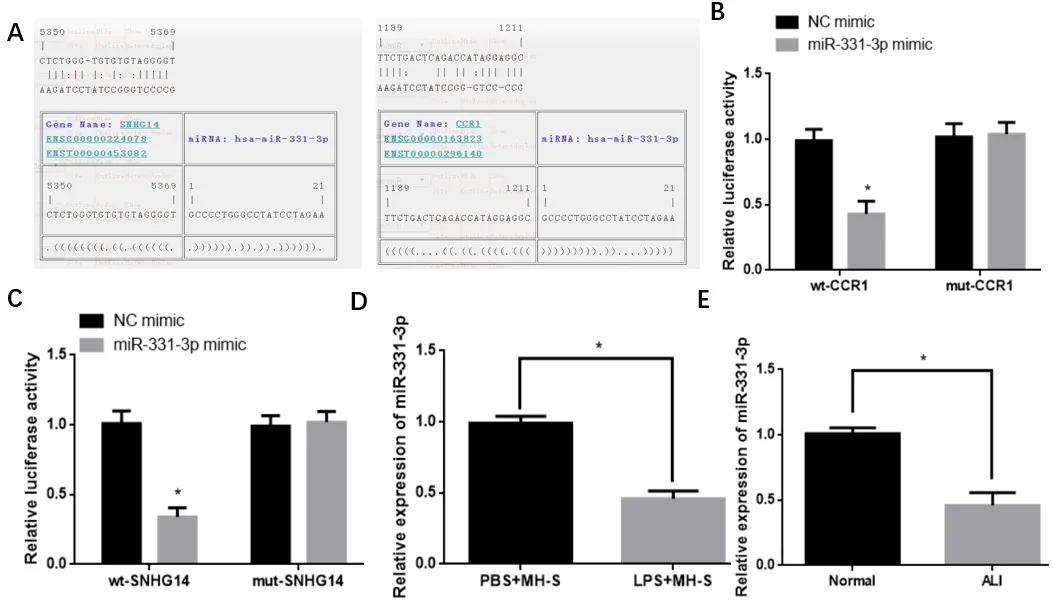
圖1 SNHG14 可能通過吸附miR-331-3p 調控CCR1 的表達
比較小鼠肺組織病理特征、肺水腫程度及肺損傷評分,并測定肺微血管通透性變化。
①HE 染色及Smith 肺損傷評分:取小鼠肺組織,10%甲醛溶液固定,梯度酒精脫水,常規石蠟包埋,切片5μm 厚。二甲苯脫蠟,乙醇復水,蘇木素染色7min,自來水沖洗,95%乙醇5s,伊紅染色1min。梯度乙醇水化,二甲苯透明,晾干,中性樹膠封片后顯微鏡下觀察組織病理學變化。同時行肺組織Smith 評分,評價肺損傷的嚴重程度。②肺濕干重比(W/D)和肺水含量測定:收集小鼠右肺下葉,測定“濕”重。然后80℃干燥肺組織,48h 后測定“干”重,肺W/D=濕重/干重。同時,根據公式計算肺水含量,肺水含量=[(濕質量一干質量)/濕質量]×100%。③比色法測定肺微血管內皮通透性變化:小鼠尾靜脈注射伊文思藍50mg/kg,將小鼠開胸取肺臟0.1-0.3g,將肺浸泡在甲酰胺溶液中,置于37℃孵育72h,將組織中色素全部浸出,取出組織,離心后取上清液,用分光光度計在620nm 波長處進行比色,根據標準曲線計算伊文思藍含量,評價肺微血管內皮通透性變化。
1.3 統計學處理
采用SPSS 21.0 統計學軟件進行數據分析,各組標準化的樣本數值用均數±標準差(±s)表示,多組間比較采用單因素方差分析,方差不齊時采用非參數的秩和檢驗,兩組之間比較采用LSD-t檢驗。P<0.05 為差異有統計學意義。
2 結果
2.1 SNHG14 可能通過吸附miR-331-3p 調控CCR1 的表達
RNA22 數 據 庫 分 析miR-331-3p 與SNHG14、CCR1 分別存在結合位點,通過雙熒光素酶驗證,與NC mimic 相比,miR-331-3pmimic 與wt-SNHG14 共轉染導致熒光強度顯著降低(P<0.05),miR-331-3p mimic 與SNHG14 被證實有靶向關系。另外,雙熒光素酶報告實驗證實,如圖1C 所示,與NC mimic 相比,miR-331-3pmimic 與wt-CCR1 共轉染導致熒光強度顯著降低(P<0.05),miR-331-3p 與CCR1 被證實有靶向關系。qRT-PCR 檢測LPS 誘導下的MH-S 細胞和小鼠肺組織中miR-331-3p 明顯低表達(P<0.05)。
2.2 SNHG14 與miR-331-3p 表達呈負相關
對miR-331-3p 與SNHG14 進行相關性分析,發現miR-331-3p 表達隨著SNHG14 表達升高而降低,并且與NC-ASO相比,SNHG14-ASO 中miR-331-3p 表達明顯升高,但是mut-SNHG14 組中miR-331-3p 表達無顯著變化,miR-331-3p 與SNHG1 呈現負相關。
3 討論
膿毒癥是宿主對感染反應失調引起的致命性器官功能障礙,而肺臟是膿毒癥時最易累及的器官,由膿毒癥引發的急性肺損傷和急性呼吸窘迫綜合征是膿毒癥患者死亡的主要原因[3]。研究表明,脂多糖是革蘭陰性菌致膿毒癥的主要抗原物質,因而通過脂多糖誘導成為模擬急性肺損傷的經典方式[2,4]。我們前期通過熒光定量PCR 檢測脂多糖誘導小鼠肺泡巨噬細胞和小鼠肺組織中lncRNA 表達情況,結果顯示SNHG14 高表達,因此,我們猜想 SNHG14 在急性肺損傷中可能發揮作用。SNHG14 在膠質瘤中,通過作為“海綿分子”抑制細胞增殖和侵襲,并促進細胞凋亡。敲除SNHG14 基因可抑制缺血性中風后釋放的炎癥細胞因子,從而對腦梗塞起保護作用,但目前尚缺乏SNHG14 在急性肺損傷中的研究[5]。本研究采用急性肺損傷經典造模術式,通過氣管內滴注脂多糖構建急性肺損傷小鼠模型,蘇木精和伊紅染色觀察肺組織病理學改變,同時,體外培養小鼠肺泡巨噬細胞,并給予終濃度為 100 μg/mL 的脂多糖刺激,培養 18 h,成功構建急性肺損傷細胞模型。
研究發現 miR-331-3p 在急性肺損傷中表達下調,認為miR-331-3p 可能是預測急性肺損傷的生物標志物。為了進一步明確miR-331-3p 在急性肺損傷中的作用機制,我們通過RNA22 數據庫及雙熒光素酶證實:miR-331-3p 與CCR1具有結合位點。趨化因子與各種炎癥反應相關,參與中性粒細胞的轉運、募集和再循環。在急性肺損傷中,趨化因子可通過增強趨化因子受體(CCR)的表達,使中性粒細胞募集至肺臟,參與了肺臟的炎癥反應[6]。在流感病毒和博萊霉素所致肺損傷模型中,CCR2 基因敲除鼠肺損傷程度明顯減輕;而CCR1 的拮抗劑可保護膿毒血癥所致肺損傷。然而對脂多糖所致肺損傷中CCRs 的調節作用卻鮮有報道[7]。我們前期通過生信分析預測SNHG14、miR-331-3p 和CCR1 具有結合位點,雙熒光素酶實驗證實它們之間具有靶向關系,且脂多糖誘導后MH-S 細胞和小鼠肺組織中miR-331-3p 表達降低,說明miR-331-3p 可能也參與了肺損傷的進程。
綜上所述,SNHG14 可能同樣作為ceRNA 吸附miR-331-3p,調控CCR1 的表達,從而參與急性肺損傷的進程。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
