Ce摻雜的NiCo2O4催化氧化甲苯性能研究
2021-01-14 03:12:34王洪華劉浩彬牛建瑞
科學技術與工程 2020年35期
關鍵詞:催化劑
王洪華,劉浩彬,牛建瑞*
(1.河北省環境科學研究院,石家莊 050000;2.河北科技大學環境科學與工程學院,石家莊 050018)
工業快速發展過程中排放大量的甲苯容易讓人產生惡心和頭痛等不適癥狀,且長期接觸可引起慢性中毒及神經系統和肝臟損傷,甚至有致癌的風險,嚴重影響了人類的生活[1]。因此尋求綠色、安全、無二次污染技術是解決甲苯問題的關鍵。與此同時,催化氧化技術可徹底礦化甲苯而被廣泛應用于甲苯處理中[2]。
催化氧化技術的核心是催化劑[3]。目前,催化材料有貴金屬、非貴金屬氧化物、復合型非貴金屬氧化物(包括鈣鈦礦型和尖晶石型等[4])。其中,Co3O4具有較好的催化氧化活性,是很有前途的VOCs催化劑。基于此研究開發了多種類型的 Co3O4催化劑,包括納米線和納米棒催化劑[5]、有序介孔Co3O4催化劑、三維有序納米Co3O4催化劑[6-8]和負載型催化劑(如以CNTs、Beta、ZSM-5、SBA-15、介孔硅為載體的催化劑[9-10])。以上CO3O4材料在催化降解VOCs上都表現出了非常優異的活性。眾所周知,較大比表面積、豐富的表面吸附氧、催化劑表面較高濃度的高價態金屬離子及較多的表面缺陷可提升催化材料活性[11],然而此類單金屬氧化物催化劑只能通過改變其物理結構來增強催化性能,且表面性質調控難以實現。
通過金屬摻雜形成雙金屬氧化物被認為是一種可調控材料表面性質的方法之一。其中通過金屬摻雜后形成尖晶石催化劑,此催化劑具有起燃溫度低、廉價、無二次污染等優勢,逐漸成為催化降解甲苯的主流催化劑。Huang等[12]研發了一種新型、低成本、高性能的3D-NiCo2O4納米片狀催化劑,用于室溫下氧化甲醛。此催化劑比表面積大,吸附有機物能力強,顯示出很高的催化氧化活性,室溫下甲醛的轉化效率可達95.3%,且連續使用200 h后催化性能基本不變。Tao等[13]采用共沉淀和熱分解法合成了NiCo2O4、Ni1.25Co1.75O4、Ni0.75Co2.25O4納米粒子,其中NiCo2O4顯示出最高的活性,在350~550 ℃溫度范圍內可完全降解甲烷。通過以上研究可以看出,NiCo2O4尖晶石在催化氧化等方面性能優異。然而,目前催化氧化性能優越的NiCo2O4[14]尖晶石多應用在電化學上,在甲苯氧化上的應用較少。強化催化劑表面活性氧生成速率有利于進一步提升其催化性能。CeO2具有較好的儲-放氧能力,能與過渡金屬之間存在特定的電子交換,且摻雜鈰可提升催化劑高價態金屬含量,改變催化材料的氧化還原性質,有利于加快催化劑表面活性氧的產生速率[15]。Wang等[16]采用Al/Ce和Al/Zr共摻雜Fe2O3催化劑,用于煤焦油的蒸汽催化裂化,摻雜后提高了輕質煤油的收率,光電子能譜(X-ray photoelectron,XPS)分析,摻雜可以增加O-的濃度,結果表明O-是決定蒸汽催化裂解的主要因素之一。
現采用水熱法制備Ce摻雜的NiCo2O4尖晶石催化劑,以甲苯為目標污染物,考察摻雜方式和摻雜量對降解甲苯效果的影響,并深入探討催化劑的穩定性及其催化甲苯機理,以期為催化氧化技術的工業化應用提供技術支持。
1 實驗部分
1.1 催化劑制備方法
1.1.1 一次水熱法制備NiCo2O4催化劑
把1 mol/L Na2CO3溶液5 mL、無水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,在集熱式恒溫加熱磁力攪拌器劇烈攪拌下以2 min的間隔分別加入,將前體溶液再攪拌20 min,將酒紅色均勻溶液轉移至高壓反應釜內,高壓反應釜在鼓風干燥箱中170 ℃水熱17 h;洗滌抽濾,在干燥箱中80 ℃下烘干200 min,烘干后在馬弗爐空氣氛圍中,以3 ℃/min升溫至300 ℃焙燒200 min得到目標催化劑。
1.1.2 一次水熱法制備Ce摻雜NiCo2O4催化劑
將1 mol/L Na2CO3溶液5 mL、無水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,Ce(NO3)3·6H2O[物質的量比為Ce(NO3)3·6H2O∶Co(NO3)2·6H2O=X(X1=5%,X2=10%,X3=15%)]等試劑在集熱式恒溫加熱磁力攪拌器劇烈攪拌下以2 min的間隔分別加入燒杯中后磁力攪拌20 min得到酒紅色溶液。將此溶液轉移至高壓反應釜內,于170 ℃水熱17 h,抽濾、洗滌數次,放置干燥箱中80 ℃下烘干。烘干后在馬弗爐空氣氛圍中,以3 ℃/min從室溫升至300 ℃焙燒200 min得到目標催化劑。
1.1.3 二次水熱法制備Ce摻雜NiCo2O4催化劑
把1 mol/L Na2CO3溶液5 mL、無水乙醇12.5 mL、1 mol/L Co(NO3)2·6H2O溶液3.4 mL、氨水11.5 mL、1 mol/L Ni(NO3)2·6H2O溶液1.7 mL,在集熱式恒溫加熱磁力攪拌器劇烈攪拌下以2 min的間隔分別加入,將前體溶液再攪拌20 min,將酒紅色均勻溶液轉移至高壓反應釜內,高壓反應釜在鼓風干燥箱中170 ℃水熱17 h;洗滌抽濾,在干燥箱中80 ℃下烘干200 min,得到目標前體物。將其溶于34.1 mL無水乙醇中,按物質的量比Ce(NO3)3·6H2O:Co(NO3)2·6H2O=X(X=15%)加入 Ce(NO3)3·6H2O,使用攪拌器攪拌20 min;將均勻溶液轉移至高壓反應釜內,高壓反應釜在鼓風干燥箱中170 ℃水熱17 h;洗滌抽濾,在干燥箱中80 ℃下烘干 200 min,烘干后在馬弗爐空氣氛圍中,以3 ℃/min升溫至300 ℃焙燒200 min得到目標催化劑。
1.1.4 二次水熱-二次焙燒法制備Ce摻雜NiCo2O4催化劑
先用1.1.1節方法制備得到一次水熱法制備NiCo2O4目標前體物,把其溶于34.1 mL無水乙醇中,按物質的量比Ce(NO3)3·6H2O∶Co(NO3)2·6H2O=X(X=15%)加入Ce(NO3)3·6H2O,使用集熱式恒溫加熱磁力攪拌器攪拌20 min;將均勻溶液轉移至高壓反應釜內,高壓反應釜在鼓風干燥箱中 170 ℃ 水熱17 h;洗滌抽濾,在干燥箱中 80 ℃ 下烘干200 min,烘干后在馬弗爐空氣氛圍中,以3 ℃/min升溫至300 ℃焙燒200 min得到目標催化劑。
1.2 催化劑表征方法
采用X射線衍射儀(D/Max-2500,Rigaku Corporation,日本)表征晶相并分析晶格參數,其中CuKα輻射(λ=0.154 06 nm)為40 kV和30 mA。對于所有催化劑,掃描角度為5°<2θ<80°,掃描速度為10 (°)/min;使用氮氣物理吸附儀(ASIQM000100-6,Quantachrome,美國)氮吸附測量樣品的比表面積測定(brunauer-emmett-teller,BET)、BJH理論方法測得孔容孔徑分布。在N2吸附之前,將樣品在473 K下脫氣4 h以除去任何殘留的水分或其他揮發物。壓力范圍p/p0=0.05~0.99;采用透射電子顯微鏡(transmission electron microscopy,TEM)(JEOL JEM 2100,日本)測量催化劑晶像及微觀形貌。X射線熒光(X-ray fluorescence,XRF)光譜儀(AXIOS,荷蘭) 基于莫斯萊定律,以特征X射線為基礎進行元素的定性、定量分析。H2程序升溫還原(H2temperature-programmed reduction,H2-TPR)裝置(AutoChem2920,美國),用電子分析天平準確稱取催化劑樣品100 mg,將稱量好的催化劑裝入U型石英管反應器中,通入N2,以10 ℃/min的升溫速率由室溫升至500 ℃,在500 ℃下對催化劑進行預處理1 h,然后溫度降至50 ℃,切換成還原氣,以H2和N2的體積比為10∶90的混合氣為還原氣,待熱導池穩定后,即可進行程序升溫還原實驗。其具體測試條件如下:還原氣體流速為30 mL/min,升溫速率為10 ℃/min,升溫至900 ℃。
1.3 催化氧化降解實驗
圖1所示為催化活性評價儀器流程,利用固定床反應器在線催化評價裝置(WFS-3015,天津先權工貿發展有限公司,中國)測試催化活性。催化劑填充粒度為40~60目,催化劑填充量為0.5 mL。通過高壓恒流泵把液態甲苯抽到汽化室,180 ℃進行汽化后,利用合成空氣吹掃甲苯至反應床層,反應后尾氣進入氣相色譜,測定甲苯濃度。進氣空速為10 000 h-1,甲苯濃度為2 538×10-6。使用HT-FFAP毛細管柱和FID檢測器,通過氣相色譜儀(GC7900,上海天美科學儀器有限公司,中國)分析甲苯的入口和出口濃度,計算去除率。甲苯的去除率為
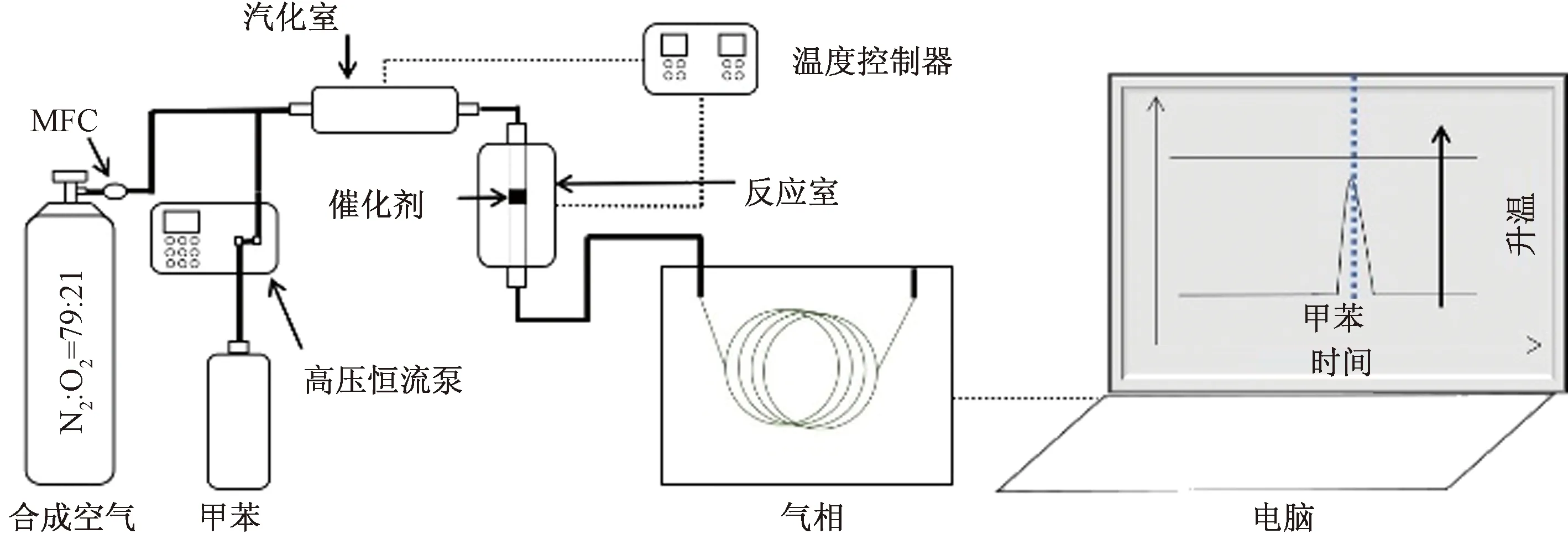
圖1 儀器流程Fig.1 Instrument flowchart
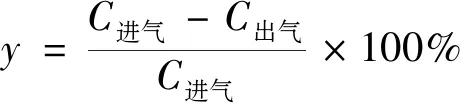
(1)
式(1)中:y為甲苯去除率,%;C進氣為氣相色譜進氣口甲苯濃度;C出氣為氣相色譜進氣口甲苯濃度。
2 結果與討論
2.1 催化劑的催化活性
圖2所示為尖晶石催化活性。使用溫度T50、T90、T99(對應于甲苯轉化率為50%、90%、99%)比較催化劑反應活性。總結圖2結果制作表1。一次水熱制備的15% Ce/NiCo2O4催化劑催化氧化甲苯顯示出最優的活性(T50=249 ℃,T90=259 ℃,T90=267 ℃),經計算得出,在267 ℃時具有最高的甲苯轉化速率[7.94 μmol/(gcats)]。比未摻雜鈰的鎳鈷尖晶石具有更高的活性,且在267 ℃下催化氧化反應72 h后催化效率僅降低3.18%。即使反應過程中有水引入,催化活性也未受到很大的影響,表明此催化劑更能適應實際工業反應過程。
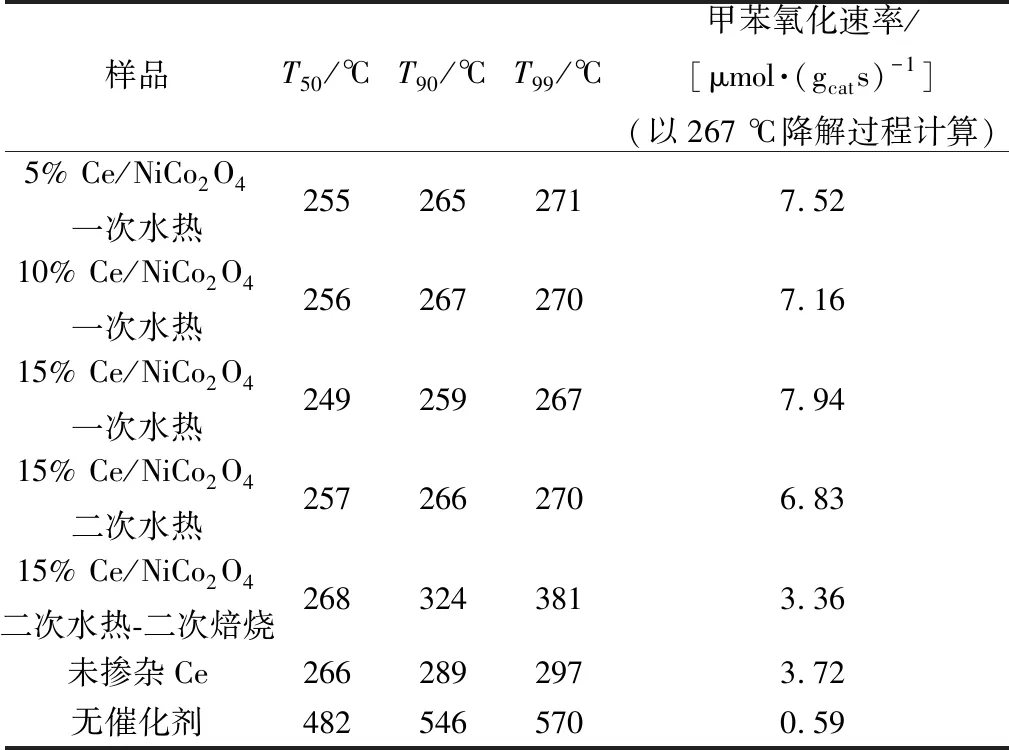
表1 T50、T90、T99對Ce/NiCo2O4的催化活性Table 1 Catalytic activities in terms of T50、T90 and T99 over Ce/NiCo2O4
2.2 催化劑的XRD分析
圖3(a)所示為水熱法制備15% Ce/NiCo2O4樣品XRD圖譜,可以看出水熱法制備15% Ce/NiCo2O4樣品均具有NiCo2O4尖晶石相(JCPDS-ICDD73-1702),在31°、36°、44°、59°、64°(2θ)處具有衍射峰,對應于(220)、(311)、(400)、(511)、(440)NiCo2O4晶面。水熱法制備的15% Ce/NiCo2O4存在CeO2相(JCPDS-ICDD89-8436),在28°、47°(2θ)處有衍射峰,對應于(111)和(220)晶面。但一次水熱法制備的15% Ce/NiCo2O4還存在CoO2相(JCPDS-ICDD70-3469),在23°(2θ)處有衍射峰,對應于(008)晶面。且一次水熱制備的15% Ce/NiCo2O4顯示最高的衍射峰強,結晶度最優,其中CoO2可能由于摻雜Ce后,部分Ce替換NiCo2O4中Co而形成的。15% Ce/NiCo2O4二次水熱過程使樣品結構坍塌,從而降低了尖晶石結晶度。而15% Ce/NiCo2O4二次水熱二次焙燒過程,樣品內部結構坍塌程度將會降低。如表2所示,由Scherrer公式計算水熱法制備的15% Ce/NiCo2O4樣品晶粒大小順序為15% Ce/NiCo2O4一次水熱法(2.40 nm)>15% Ce/NiCo2O4二次水熱法(1.967 nm)>15% Ce/NiCo2O4二次水熱-二次焙燒法(1.367 nm)。水熱法制備的15% Ce/NiCo2O4樣品晶胞尺寸相同均為 0.534 nm3,表明樣品屬于立方體晶系。
圖3(b)所示為水熱法制備xCe/NiCo2O4(x=5%、10%、15%)樣品XRD圖譜,可以看出水熱法制備xCe/NiCo2O4(x=5%、10%、15%)樣品均具有NiCo2O4尖晶石相(JCPDS-ICDD73-1702),在31°、36°、38°、44°、59°、64°(2θ)處具有衍射峰,對應于(220)、(311)、(222)、(400)、(511)、(440)NiCo2O4晶面。當摻雜量為5% Ce/NiCo2O4時,存在少量的CoO2相(JCPDS-ICDD70-3469),在23°(2θ)處有衍射峰,對應于(008)晶面。隨著摻雜量的增加,10%Ce/NiCo2O4時,CoO相衍射峰的峰強明顯增加,表示CoO的量增加而CoO2相消失。當15%Ce/NiCo2O4時,CoO相衍射峰基本消失,出現了CeO2相衍射峰(JCPDS-ICDD89-8436),在28°和47°(2θ)處有衍射峰,對應于(111)和(220)晶面,表明摻雜已經處于飽和狀態。且隨著Ce摻雜量的增加,催化劑中CoO2含量也明顯上升,有效提高了高價態金屬含量,且通過對一次水熱、二次水熱以及二次水熱二次焙燒條制備的鎳鈷尖晶石進行氫氣程序升溫還原發現,一次水熱制備的材料其還原峰向低溫偏移,表明一次水熱制備的催化劑具有更好的氧化還原性質,有利于活性氧的產生,對于高效催化氧化降解甲苯具有重要作用。同時晶粒計算結果如表2所示,由Scherrer公式計算水熱法制備xCe/NiCo2O4(x=5%、10%、15%)樣品晶粒大小順序為10% Ce/NiCo2O4(3.70 nm)>5% Ce/NiCo2O4(2.767 nm)>15% Ce/NiCo2O4(2.40 nm)。較小的晶粒有利于催化過程中和目標污染物更好地接觸,進一步提升催化反應效率。水熱法制備的xCe/NiCo2O4(x=5%,10%,15%)樣品晶胞體積相同均為0.534 nm3,表明樣品屬于立方體晶系。
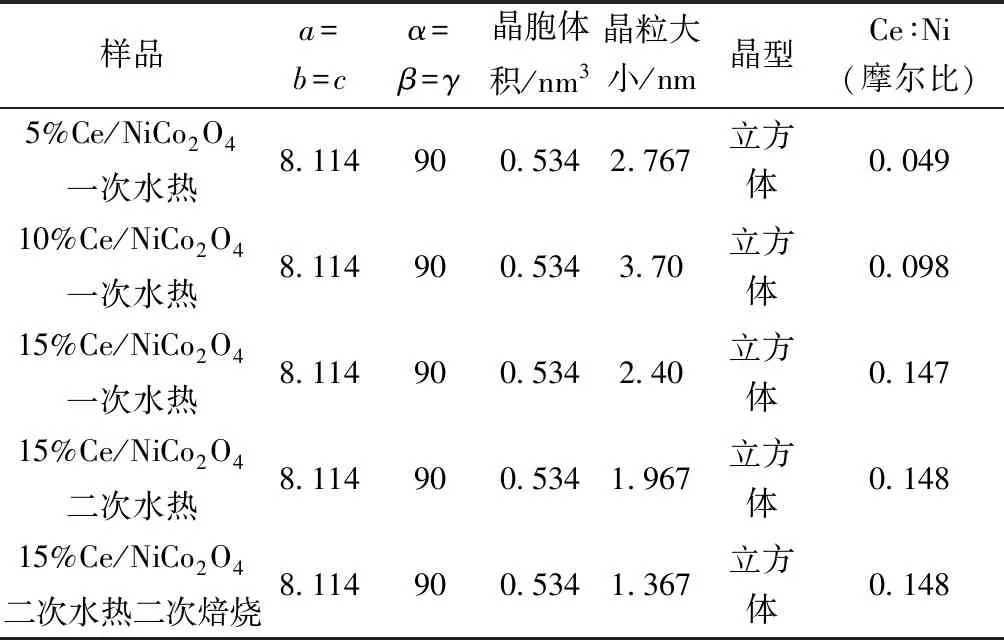
表2 xCe/NiCo2O4(x=5%,10%,15%)樣品XRD及XRF數據Table 2 XRD and XRF data of xCe/NiCo2O4(x=5%,10%,15%)
2.3 催化劑的BET分析
圖4所示為xCe/NiCo2O4(x=5%、10%、15%)為N2吸脫附曲線和孔徑分布,可以看出xCe/NiCo2O4(x=5%,10%,15%)屬于典型的介孔分布曲線,BJH理論方法測得主要孔徑分布在4 nm左右,如表3所示,xCe/NiCo2O4(x=5%,10%,15%)孔容BJH理論方法測得主要集中在0.1 cm2/g。從N2吸脫附曲線可知,遲滯環曲線屬于H3型遲滯環回歸線,在高壓力區域沒有表現出任何吸附限制。其中一次水熱15% Ce/NiCo2O4在高壓區域和低壓區域都沒有表現出任何吸附限制,表明該催化劑孔道結構均勻有序,可以對N2優先進行多層吸附。
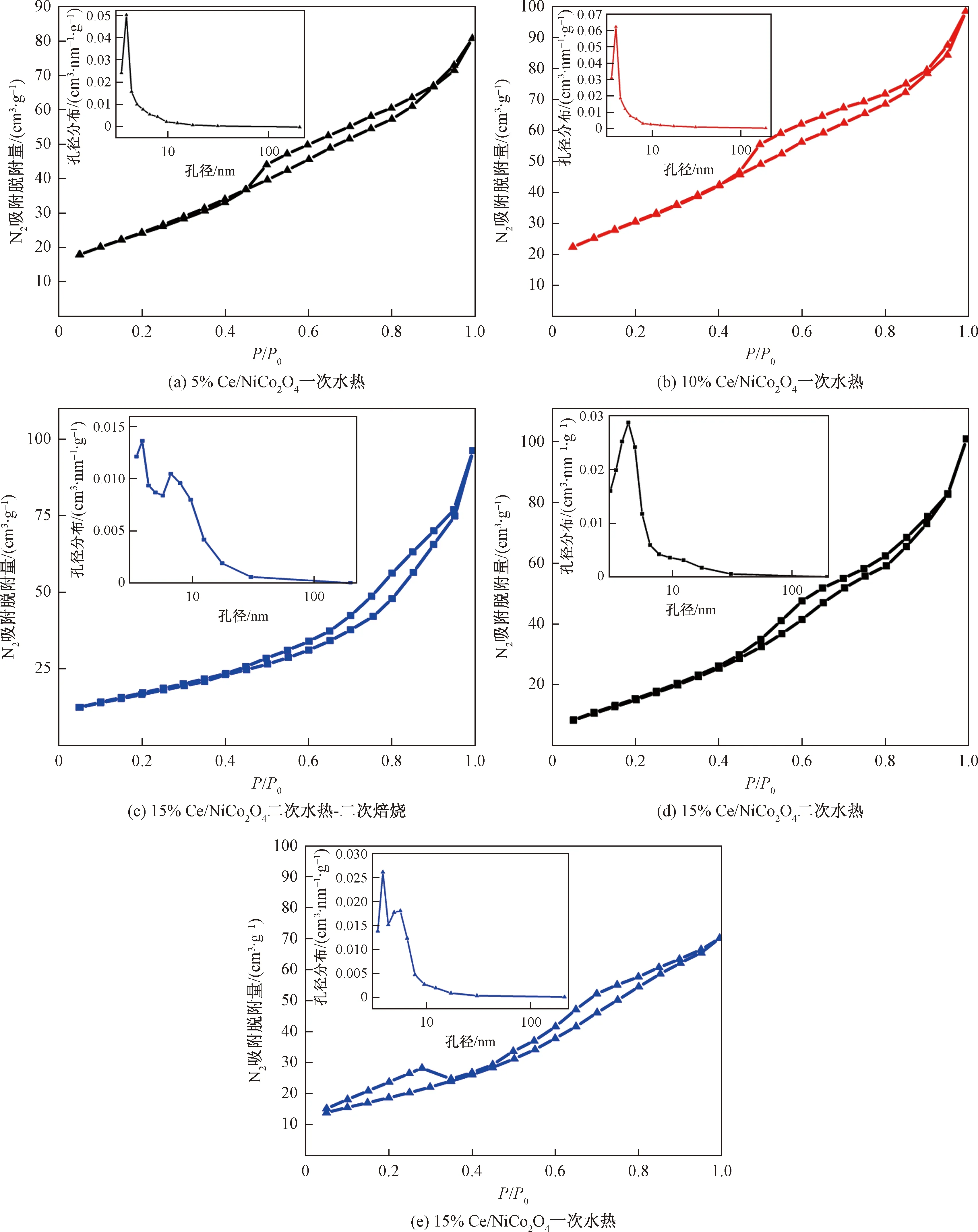
P代表氣體真實壓力;P0代表氣體在測量溫度下的飽和蒸汽壓;P/P0代表相對壓力圖4 xCe/NiCo2O4(x=5%,10%,15%)比表面積和孔容-孔徑分布Fig.4 N2 adsorption-desorption isotherms and pore size distribution of xCe/NiCo2O4(x=5%,10%,15%)
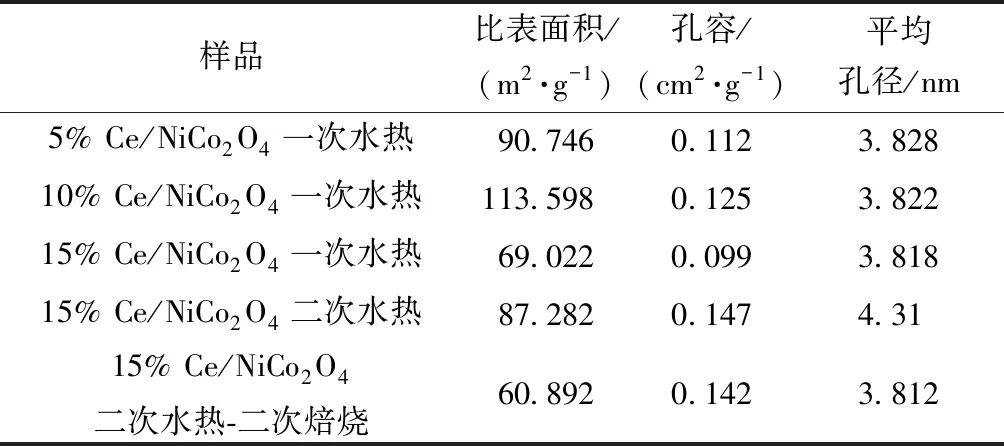
表3 材料氮氣物理吸附結果Table 3 Textural properties of as-prepared materials
2.4 催化劑的TEM分析
圖5所示為一次水熱制備的15% Ce/NiCo2O4尖晶石催化劑TEM圖。由圖5(a)可以看出15% Ce/NiCo2O4形貌為棒狀,從圖5(b)、圖5(c)可以看出棒狀主要由顆粒堆積組成。圖5(d)、圖5(e)晶格尺寸分別為0.50 nm和0.28nm,對應的晶像分別為(111)和(220)。圖5(f)為15%Ce/NiCo2O4衍射圖。表明通過一次水熱法,摻雜Ce可以得到形貌小顆粒堆積而成的棒狀。

圖5 15% Ce/NiCo2O4一次水熱的TEM圖和衍射圖Fig.5 TEM and diffraction pattern of 15% Ce/NiCo2O4 after hydrothermal reaction
2.5 催化劑的催化氧化機理
如圖6所示,采用一次水熱摻雜Ce到NiCo2O4中制備尖晶石催化劑,隨著摻雜量的增加,NiCo2O4中Ce達到飽和后以多余的CeO2形式存在。在氧化還原過程中,高價態的Co被還原,釋放出活性氧原子即表面吸附氧,活性氧一部分參與反應,多余的則會儲存在CeO2上。當甲苯達到催化劑表面時,表面活性氧起到主要降解作用,CeO2可通過自身氧化還原過程補充被逐漸消耗的活性氧,最終保持催化活性將甲苯有效分解為CO、H2O和CO2。圖7所示為質譜數據,反應后未出現甲苯的質譜信號,表明已被完全降解。同時由于高價金屬釋放活性氧之后形成空穴,空氣中氧會被空穴活化后重新形成活性氧,因此高價態金屬含量越高,氧化還原性質越好,越有利于活性氧的生成。摻雜Ce主要有兩方面作用:①制備具有較好的儲-放氧特性的催化劑;②Ce的摻雜可提升催化劑中高價態金屬含量,為活性氧的產生提供良好的條件。
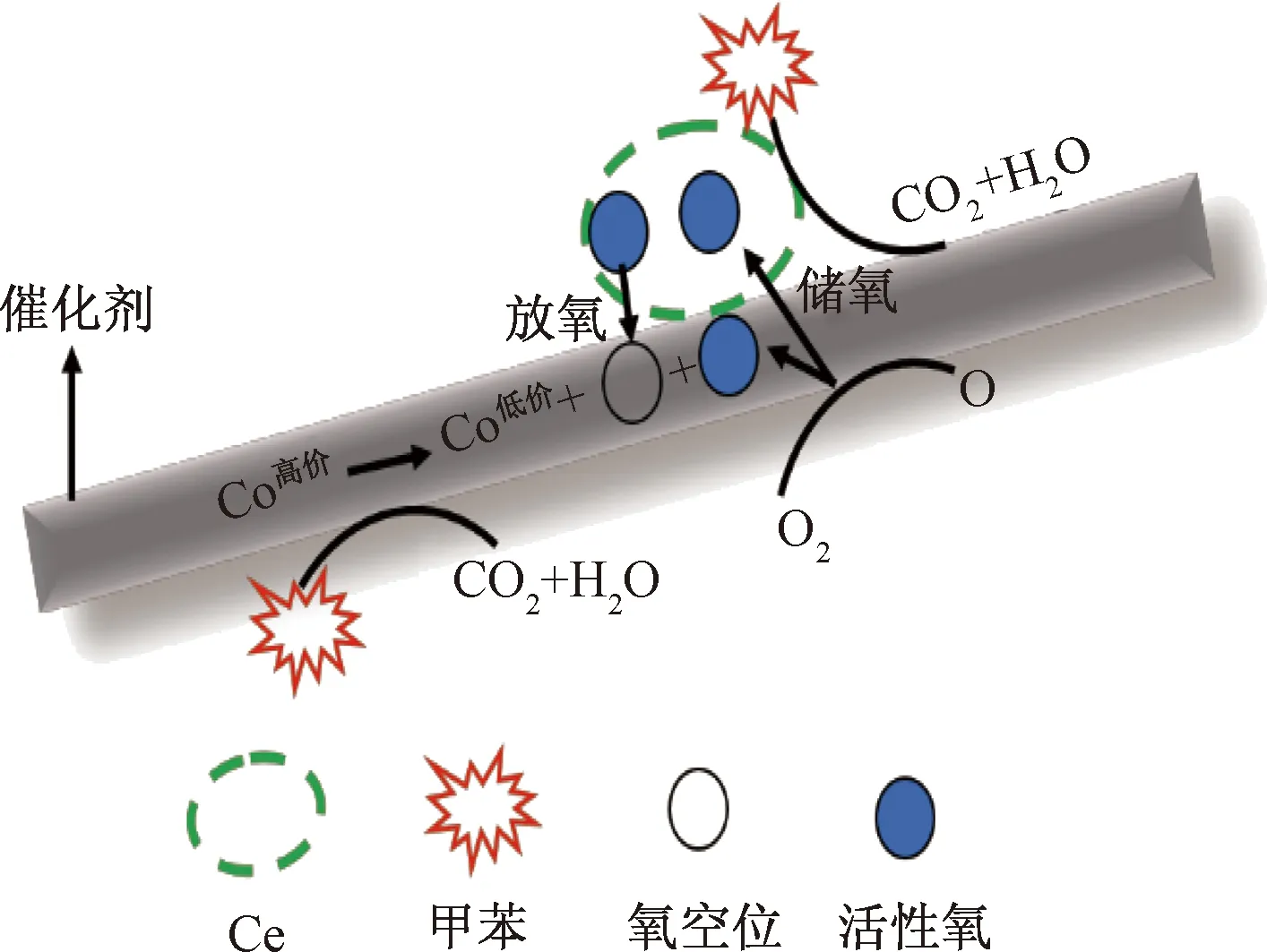
圖6 催化氧化機理Fig.6 Mechanism of catalytic oxidation
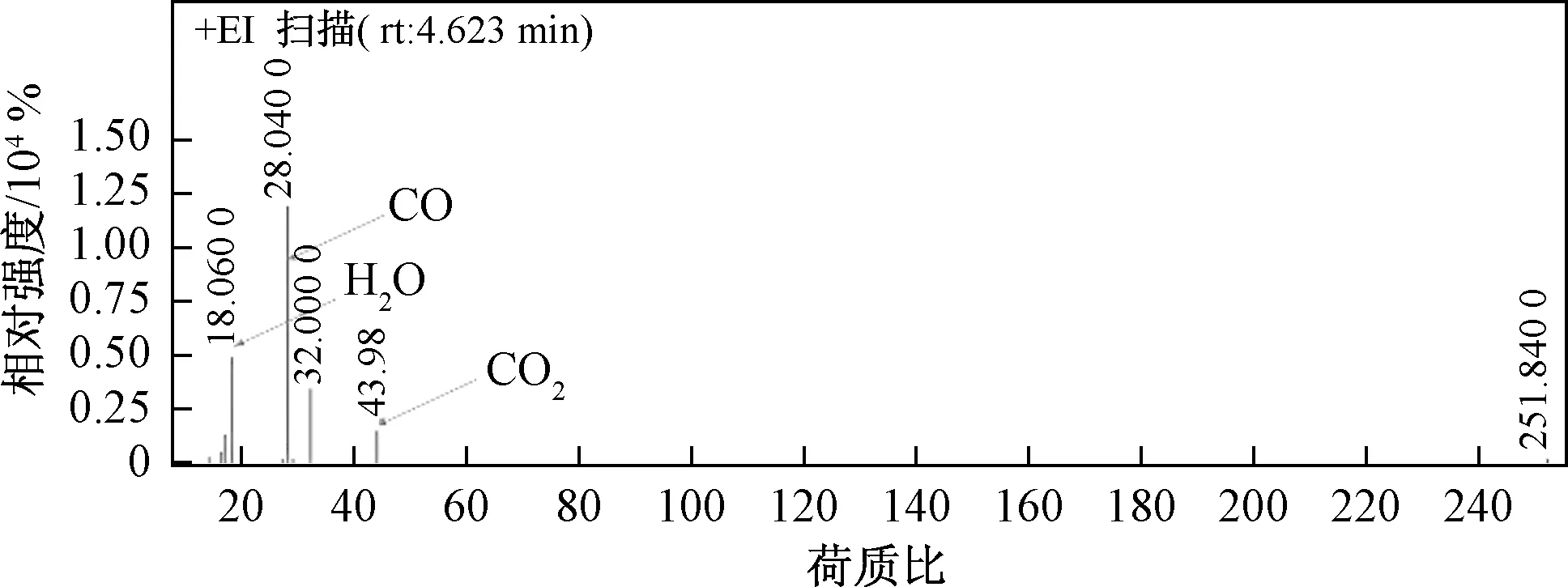
圖7 質譜數據Fig.7 The data of GC-MS
3 結論
通過水熱法將Ce摻雜到NiCo2O4中,并探究摻雜量對材料性能的影響,得出以下結論。摻雜Ce可以提高NiCo2O4中氧的生產速率。以甲苯為目標污染物,甲苯濃度為2 538×10-6,空速為10 000 h-1,催化劑體積為0.5 mL,一次水熱15% Ce/NiCo2O4表現出最優的催化氧化性能,一次水熱15% Ce/NiCo2O4的催化溫度為T50=249 ℃、T90=259 ℃、T90=267 ℃;通過在267 ℃下連續催化氧化72 h后催化劑的催化氧化性能無顯著變化,且引入5%水后對催化性能也沒有產生任何影響,表明一次水熱15% Ce/NiCo2O4催化劑具有良好的穩定性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
