基于生物信息學方法篩選乙型肝炎病毒相關肝癌的關鍵基因和臨床預后
2021-01-14 07:15:06李玉杰楊雪佳吳登強呂玥茜李茵佳周素芳
海南醫學 2020年24期
李玉杰,楊雪佳,吳登強,呂玥茜,李茵佳,周素芳
廣西醫科大學基礎醫學院,廣西 南寧 530021
肝細胞癌是一種常見的惡性肝病[1],HCC的死亡率在消化系統腫瘤中居第三位[2-5]。在大多數情況下,病毒感染導致HCC的發生、侵襲和轉移,是一個全球性的公共衛生問題[6-7],特別是慢性乙型肝炎病毒(HBV)是導致HCC的主要原因之一[8]。超過50%的肝癌病例是由慢性HBV感染引起的,在高流行地區,HBV感染估計占肝癌病例的80%以上[3]。此外,HBV相關肝癌患者的轉移和復發率明顯高于未感染HBV的患者,HBV是證明與HCC發展直接相關的首批病毒之一[2,4,9]。目前可用的抗病毒藥物并不能完全消除慢性乙肝病毒[10-11],慢性HBV感染可導致肝臟慢性炎癥[12-13],從而導致正常肝細胞轉化為癌細胞,這使得乙肝病毒和煙草成為最重要的環境致癌物質。雖然HBV與HCC之間的因果關系已經確立,但肝癌發生的原因和潛在機制仍有待于充分闡明[6,11,13]。后基因組技術的出現以及全球公開數據集的顯著發展,為研究肝癌的發病機制提供了工具,使得研究在單個陣列中同時表達數千個基因的mRNA成為可能,利用生物信息學方法可以識別與腫瘤消退相關的潛在生物標志物和信號通路[14]。最近,許多研究使用全基因組數據集來識別肝癌的診斷和預后分子標記,特別是來自癌癥基因組圖譜(TCGA,The Cancer Genome Atlas)研究網絡和基因表達綜合數據庫(GEO)的HCC數據集。人類HCC的基因表達分析已經成功地根據預后、病因和肝內復發對HCC進行了分子分類[3,15-16]。但是目前利用微陣列技術對HBV相關的HCC研究較少[17],相關的生物標記物較多,不能為臨床預后提供針對性的依據,迫切需要進一步對相關的全基因組數據進行全面的基因組分析,以研究診斷和預后的生物標志物[4,18]。在本研究中,根據GEO和TCGA的數據,通過對HBV相關的肝癌組織和非腫瘤組織差異表達基因(DEGs)的研究,還對DEGs進行了基因本體(GO)功能富集分析和基因組京都百科全書(KEGG,The KEGG resource for deciphering the genome)途徑富集分析,此外,還有蛋白質-蛋白質相互作用(PPI)網絡等,分析了基因表達與HCC預后和進展的關系。本研究的結果有助于確定肝癌患者個體化治療和乙肝病毒感染史的關鍵生物標記物,為進一步了解腫瘤進展和肝癌的研究提供依據[19-21]。
1 材料與方法
1.1 微陣列數據篩選 首先從基因表達數據庫(GEO,https://www.ncbi.nlm.nih.gov/geo/)下 載 基 于GPL570(HG-U133_Plus_2)人類基因組U133 Plus 2.0陣列的mRNA表達數據集GSE55092和GSE121248,GSE55092包括49個HBV相關的HCC樣本和91個非腫瘤樣本,GSE55092包括70個HBV相關的HCC樣本和37個非腫瘤樣本,使用GEO2R在線分析工具(https://www.ncbi.nlm.nih.gov/geo/geo2r/)對每個數據集進行計算,調整后P<0.01和表達倍數變化值|log2(fold change)|≥2的基因被認為是差異表達。“gplots”R軟件包用于篩選由2個微陣列數據集的DEGs列表,通過韋恩圖(http://bioinformatics.psb.ugent.be/webtools/Venn/)分析共同包含的差異表達基因[22-23]。
1.2 DEGs功能和通路分析 注釋可視化與綜合發現數據庫(DAVID 3https://DAVID.ncifcrf.gov/home.jsp;6.8版)是一個生物信息數據庫,它集成了生物數據和分析工具,為大規模基因的生物功能提供系統全面的注釋信息,蛋白質列表幫助用戶從中提取生物信息[24]。KEGG是世界上最常用的了解高級功能和生物系統的生物信息數據庫之一[25]。在分子水平上,KEGG整合了由高通量實驗技術產生的大量實用數據庫資源。GO被廣泛應用于生物信息學中,包括細胞成分(CC)、分子功能(MF)和生物過程(BP)三個方面[18]。為了分析DEGs的GO和通路富集信息,使用了DAVID在線工具,P<0.05為差異具有統計學意義。
1.3 PPI網絡構建與關鍵基因篩選 檢索相互作用的基因和蛋白質的搜索工具string(https://string-db.org/;11.0版),是一個用于識別DEG之間相互作用和功能關聯的系統,能夠構建蛋白質-蛋白質相互作用(PPI)網絡。在Cytoscape 3.5.1(https://cytoscape.org/)軟件中,利用CytoHubba插件獲得PPI網絡中得分最高的前14個hub基因。
1.4 遺傳標記的生存分析和臨床特點 GEPIA(Gene Expression Profiling Interactive Analysis,http://gepia.cancer-pku.cn/)是一個新開發的交互式網絡服務器,用于分析9 736個腫瘤和8 587個正常樣本的RNA序列表達數據,通過GEPIA分析hub基因在腫瘤不同階段的表達量和在不同腫瘤中的表達差異,利用cBio-Portal(http://www.cbioportal)分析來自TCGA的hub基因之間的相關性。使用Kaplan-Meier Plotter(www.kmplot.com)的Kaplan-Meier曲線特征分析與hub基因相關的總體生存率,其中包括364例患者。
1.5 統計學方法 GEO數據集的校正P值用GEO2R軟件計算,調整P<0.01的數據被認為差異有統計學意義。GO項和KEGG途徑P<0.01,被認為顯著富集。對于Kaplan-Meier分析,生存曲線比較采用對數秩檢驗,P<0.05為差異有統計學意義。
2 結果
2.1 DEGs的篩選 利用GEO2R在線分析工具處理HCC表達微陣列數據。基于|logFC|>2.0和校正P<0.01篩選差異表達基因的層次聚類熱圖,從GSE55092數據集中共篩選出389個DEGs,聚類熱圖如圖1A所示,GSE121248篩選出來了146個DEGs,聚類熱圖如圖1B所示,VENN圖中顯示兩個數據集中包含了129個DEGs如圖1C。
2.2 GO功能注釋和KEGG富集分析 肝癌中的DEGs的GO功能注釋和KEGG富集分析的主要功能和通路分別在圖2A~2D中顯示。GO中的BP顯著富集為“氧化還原過程”、“外源藥物分解代謝過程”和“一元羧酸代謝過程”,CC主要表現為“細胞外區域”和“細胞器膜”,MF主要富集在“血紅素結合”、“鐵離子結合”和“分子氧的結合或還原”,KEGG顯示DEGs在“視黃醇代謝”、“咖啡因代謝”和“p53信號通路”中富集程度最高。目前的結果表明,DEGs在細胞分裂和代謝途徑方面有顯著的富集作用。
2.3 DEGs的PPI網絡分析 共有129個DEGs用于構建PPI網絡,如圖3A所示,根據CytoHubba插件中的最大集團中心性算法(MCC,maximal clique centrality)算法篩選出前14個hub基因,細胞周期蛋白依賴性激酶1(CDK1)[26]、細胞周期蛋白B1(CCNB1)、DAN拓撲異構酶(TOP2A)、重組人核糖核苷酸還原酶M2(RRM2)、上皮細胞轉化序列2癌基因(ECT2)、微管相關蛋白(PRC1)、透明質酸介導運動因子受體重組蛋白(HMMR)、異常紡錘型小腦畸形癥蛋白基因(ASPM)、泛素蛋白連接酶(DTL)、人類重組蛋白(RACGAP1)、微小RNA-217靶基因(ANLN)有絲分裂檢查點絲氨酸/蘇氨酸激酶B(BUB1B)、PDZ結合激酶(PBK)、細胞周期相關蛋白激酶 2(NEK2)。在這 14 個 hub 基因中(圖 3B),CDK1、CCNB1和TOP2A顯示較高的得分,其名稱、縮寫和功能見表 1[15,27-28]。
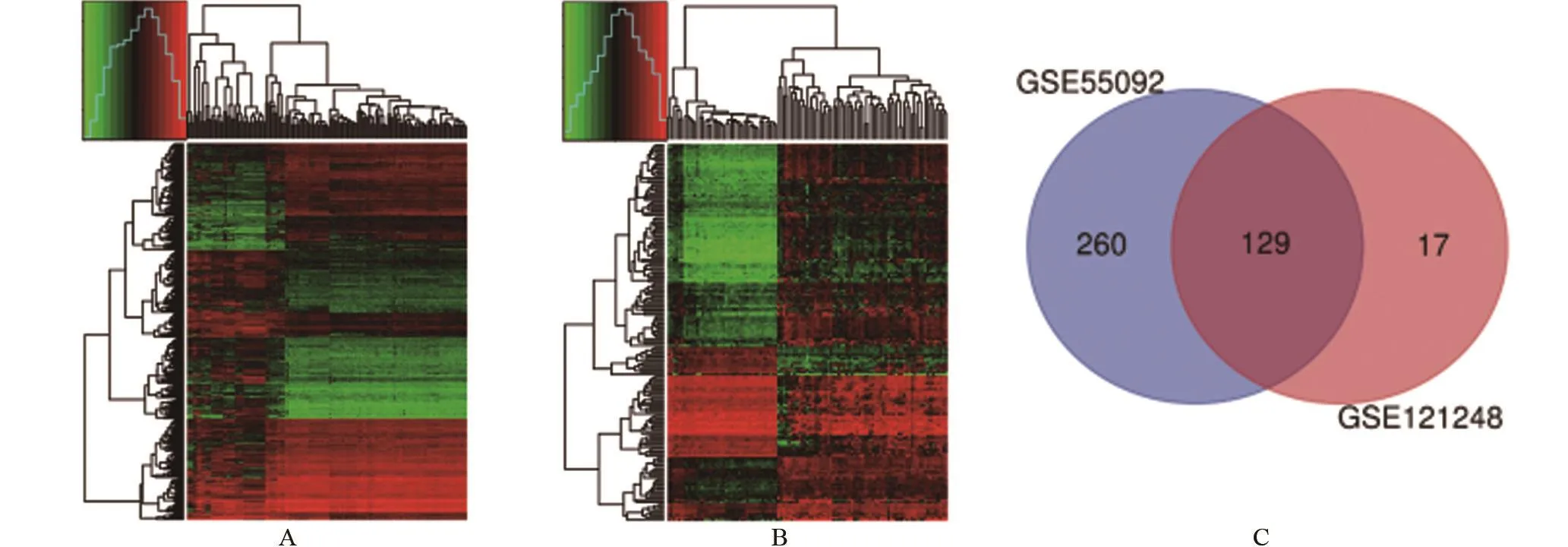
圖1 基于|logFC|>2.0和校正P值<0.01篩選差異表達基因的層次聚類熱圖和韋恩圖

圖2 對DEGs進行GO和KEGG通路富集分析
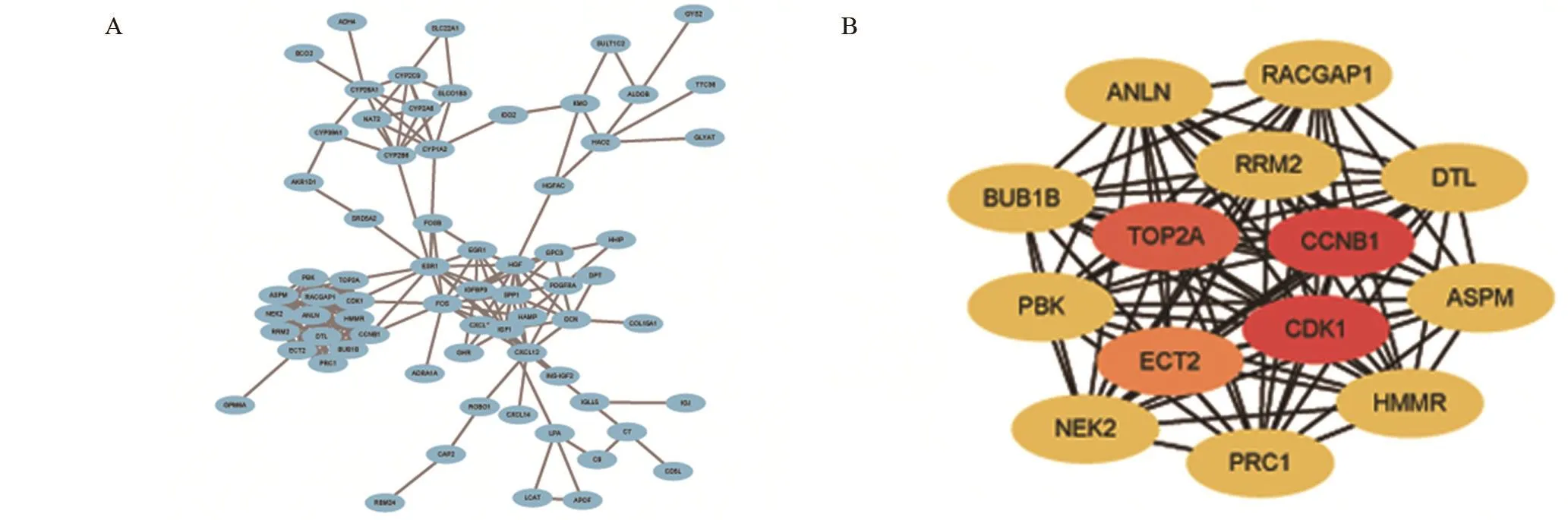
圖3 DEGs與前10位hub基因的PPI網絡構建

表1 關鍵基因的功能信息
2.43 種HCC遺傳標記的預后潛力和相關性分析 根據cBioPortal數據庫(圖4A~4C),3個hub基因CDK1、CCNB1和TOP2A之間存在顯著相關性(P<0.05),利用GEPIA數據集比較了HCC和肝組織中CDK1、CCNB1和TOP2A的mRNA表達。結果表明,hub基因在肝癌組織中的表達水平均高于正常組織(圖4D~4F),緊接著分析hub基因在肝癌分期中的表達,結果差異無統計學意義(圖5A~5C)。通過分析CDK1、CCNB1和TOP2A表達對肝癌患者生存和預后的影響[16],Kaplan-Meier曲線和對數秩檢驗分析顯示,CDK1、CCNB1和TOP2A水平的升高與肝癌患者的總體生存率(OS)降低顯著相關,高表達的輕微趨勢與預后不良相關(P<0.05;圖5D~5F)。
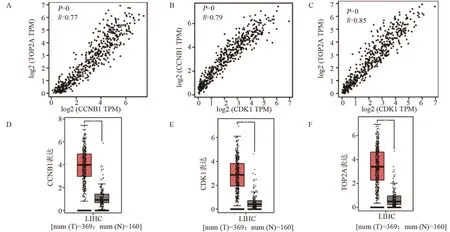
圖4 hub基因相關性和表達量分析
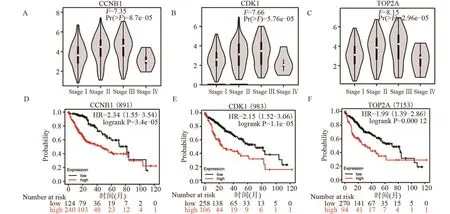
圖5 hub基因在不同腫瘤分級中的表達和生存分析
3 討論
肝癌的發生是一個復雜的生物學過程[1]。近年來,大量的生物標志物被用于肝癌的早期診斷[29],尤其是慢性乙肝病毒是導致HCC的重要原因之一,但是現在關于此的研究還很少,多基因水平的研究有助于探索癌癥的發病機制[8,19,30]。本研究采用生物信息學方法對2個肝癌基因芯片的數據進行分析,最后發現CDK1、CCNB1和TOP2A與患者生存率低、腫瘤分級高有關。本研究主要針對HBV感染的肝癌病例,有別于以往的研究[3,7,10,12,31-32],本研究還分析了臨床特征與生物標志物的關系,這些基因的表達與總生存率和病理分期有關[13]。為了探討肝癌的分子機制,在兩個來自GEO數據庫的數據集中,共有139個HBV感染的腫瘤樣本,128個非腫瘤樣本,通過GEO2R和韋恩圖分析共得到129個DEGs,經過富集和PPI分析,共鑒定出了14個hub基因。在這14個hub基因中,CDK1、CCNB1和TOP2A相互作用密切,KEGG分析表明,在“視黃醇代謝”、“咖啡因代謝”和“p53信號通路”中富集程度最高,說明DEGs顯著影響細胞分裂和代謝途徑等方面,促進后期復合物依賴性分解代謝過程、氧化還原過程和細胞分裂,是前3位富集最顯著的BPs,腫瘤發生的主要原因可能是細胞周期失衡,進而導致細胞過度增殖。先前有相關研究報道,HBV感染可引起代謝信號通路的改變[2,6,8,20,31],其結果可能改變正常的肝細胞代謝,從而有助于乙肝相關致癌的進展[6]。
CDK1是CCNB1在G2/M轉換和有絲分裂恢復中所必需的[33],兩者是高度相關的,HBV可以激活HCC細胞中的CCNB1-CDK1激酶。在其他研究中,發現HBV陽性患者的HCC組織中CCNB1和CDK1上調。并且,這兩個基因的過度表達與預后不良有關。CDK1被認為是CCNB1的重要組成部分,因為它可以影響HBV陽性肝癌患者的總生存率和無復發生存率。TOP2A過表達與肝癌早期發展、短期生存和化療耐藥有關。在本研究中,利用TCGA的數據進一步分析表明,CDK1、CCNB1和TOP2A之間存在密切的相關性,3個hub基因在肝癌組織中的表達明顯高于癌旁組織,三者與肝癌患者的生存率顯著相關,HCC組織中CDK1、CCNB1和TOP2A的高表達與預后差和復發風險高有關。然而,這3個基因在肝癌和其他癌癥類型中的生物學功能,包括代謝和細胞周期,尚需進一步研究,這可能是它們作為治療肝癌或者其他癌癥類型的靶點。
總之,本研究在一個大的隊列基礎上,與HBV感染患者的非腫瘤組織相比,在HCC組織中發現了幾個DEG。基于DEGs,確定了幾個關鍵途徑。同時,PPI網絡也揭示了這些途徑中DEGs的相互作用。一些結果與之前的研究一致[20]。本研究為有HBV感染史的患者提供了新的HCC病因和非腫瘤肝組織轉化為HCC組織的分子機制。重要的是,這些結果可能為這些患者的靶向治療提供一些潛在的治療靶點,有助于肝癌的早期診斷和治療。
猜你喜歡
天天愛科學(2022年9期)2022-09-15 01:12:54
天天愛科學(2022年4期)2022-05-23 12:41:48
當代水產(2022年3期)2022-04-26 14:26:56
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
航空世界(2020年10期)2020-01-19 14:36:20
天津醫科大學學報(2019年3期)2019-08-13 06:53:08
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
腫瘤預防與治療(2015年1期)2015-09-26 07:26:20
中國當代醫藥(2015年16期)2015-03-01 02:03:11
