成纖維樣滑膜細胞能量代謝異常關鍵分子在類風濕關節炎中的研究進展
2021-01-26 06:13:50賈成艷
中國藥理學通報 2021年1期
賈成艷,常 艷,魏 偉
(安徽醫科大學臨床藥理研究所,抗炎免疫藥物教育部重點實驗室,抗炎免疫藥物安徽省協同創新中心,安徽 合肥 230032)
類風濕關節炎(rheumatoid arthritis,RA)是一種全身性自身免疫病,主要累及關節滑膜,產生慢性滑膜炎癥,最終導致關節破壞和功能喪失。該病的病理機制涉及多種類型細胞之間的相互作用,其中成纖維樣滑膜細胞(fibroblast-like synoviocytes,FLS)起關鍵作用,表現出一種侵襲性表型,產生炎癥介質導致滑膜炎癥,造成軟骨、骨損傷。隨著代謝組學的發展,細胞代謝研究為RA的治療提供了潛在新靶點。雖然,目前對于RA的治療已經取得一定的進展,但缺乏直接針對FLS代謝異常的藥物。越來越多的證據表明,干擾RA-FLS代謝途徑中的某些分子,可以緩解RA疾病嚴重程度,一些關鍵的轉運體和代謝酶成為潛在的藥物治療靶點。
1 FLS能量代謝異常介導了RA的病理機制和發生發展
FLS是滑膜組織的主要組成細胞,具有分泌炎性細胞因子及趨化因子促進炎癥反應、分泌基質金屬蛋白酶(matrix metalloproteinase,MMP)降解軟骨、刺激破骨細胞分化導致骨侵蝕等功能。此外,由于表面標記物(Tab 1)的表達差異,不同功能FLS表型的發現[1-2],進一步闡明了FLS在RA病理機制中的作用。
近年來,對RA代謝途徑的研究改變了對自身抗原識別引發自身免疫的早期觀點。RA患者血清中糖酵解產物和氨基酸代謝產物增加,且脂質代謝產物與C-反應蛋白水平相關,提示RA患者能量代謝發生異常[3]。RA患者的原始T細胞代謝紊亂,戊糖磷酸途徑活性增強,產生高水平的還原型煙酰胺腺嘌呤二核苷酸磷酸和生物合成前體,降低了細胞內活性氧水平,促進Th1和Th17的分化。葡萄糖轉運體1(glucose transporter 1,GLUT1)表達增強,增加葡萄糖攝取及糖酵解通量,提供細胞所需能量,增強M1巨噬細胞及B細胞活性;線粒體脂質氧化途徑可以刺激M2巨噬細胞及記憶B細胞功能[4]。RA-FLS異常活化具有侵襲性,介導炎癥和關節破壞,代謝需求高。研究表明RA-FLS中糖酵解途徑高度激活,脂質(如膽堿、花生四烯酸)及氨基酸(如色氨酸、谷氨酰胺)的代謝發生改變,這些代謝變化參與了FLS的異常活化和滑膜炎癥。
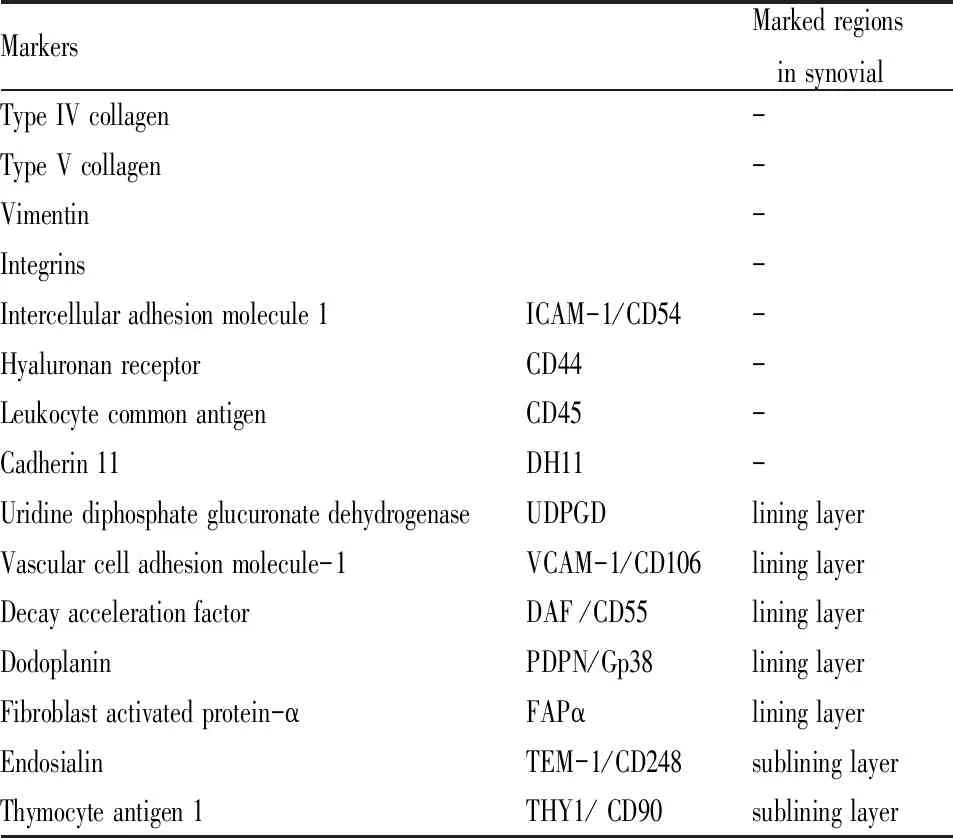
Tab 1 Surface markers of FLS
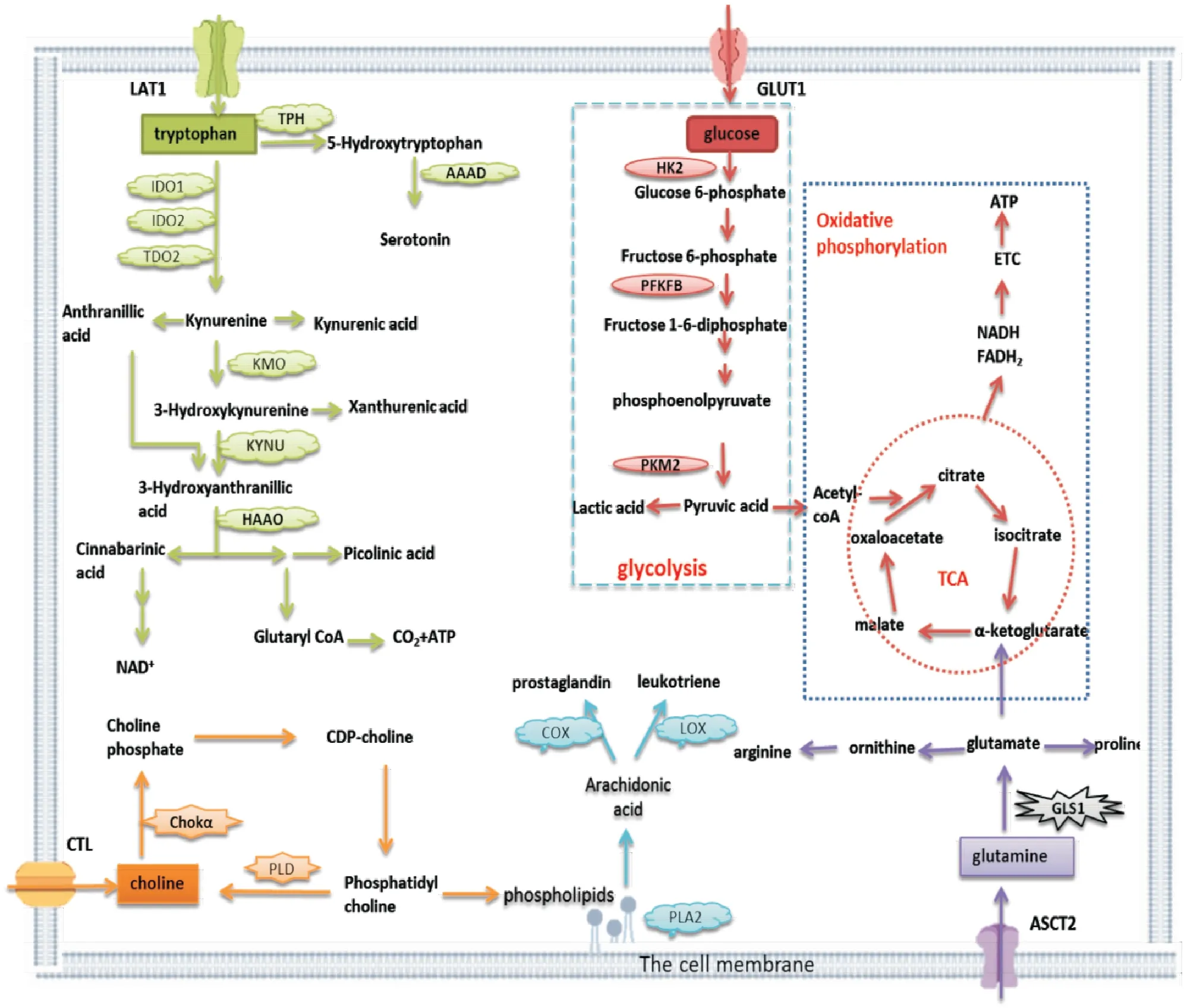
Fig 1 The metabolic pathway of RA-FLS
臨床上用于治療RA的藥物可以通過影響代謝發揮治療作用。甲氨蝶呤(methotrexate,MTX)作為治療RA患者的一線藥物,影響葉酸代謝,抑制DNA的生物合成及FLS增殖。并且,MTX能顯著降低RA-FLS中己糖激酶II(hexokinases 2,HK2)的表達,調控FLS糖酵解活性。甾體抗炎藥糖皮質激素能調節許多與糖酵解和雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路相關的代謝酶的基因轉錄,從而發揮治療RA的作用[5]。以上結果提示,調控RA異常代謝恢復其正常免疫穩態可能是治療RA的潛在新策略。
2 FLS能量代謝途徑及異常關鍵分子
葡萄糖、脂質、氨基酸代謝途徑中異常活化的轉運體及代謝酶,導致FLS能量代謝異常,可能是靶向能量代謝異常細胞的關鍵分子(Tab 2)。抑制FLS代謝途徑中異常關鍵分子的活化,具有潛在的治療關節炎的作用,對RA藥物的開發具有重要作用。
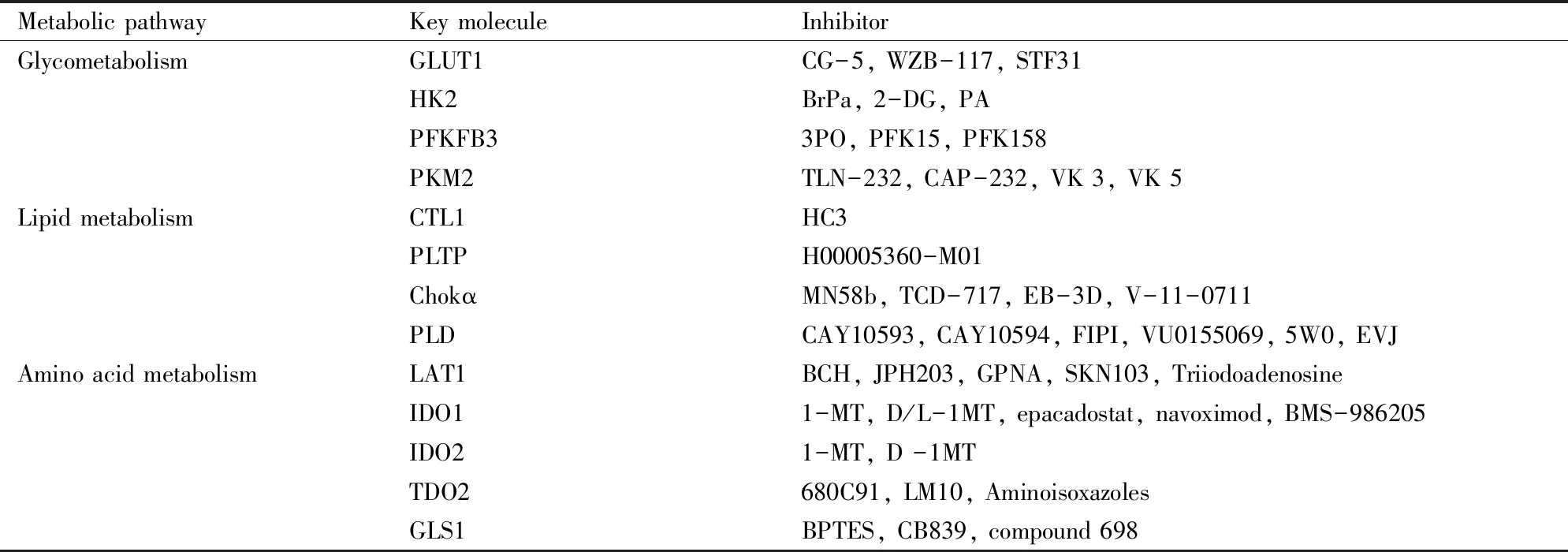
Tab 2 The key molecule of abnormal energy metabolism in RA-FLS
2.1 糖酵解代謝途徑
2.1.1糖酵解代謝途徑轉運體 研究表明[6],RA-FLS中GLUT1表達增加,并且與HK2、MMP-3表達水平相關。將RA患者CD4+T細胞與RA-FLS體外共培養后,CD4+T細胞分泌促炎因子的能力增強,RA-FLS中GLUT1、GLUT3、MMP-3、MMP-9表達增加,導致RA-FLS侵襲性及糖酵解活性增強,糖酵解抑制劑2-DG可逆轉RA-FLS上述反應。動物實驗發現,K/BxN關節炎小鼠FLS中GLUT1表達增加并促進FLS遷移及分泌MMP-3的能力,糖酵解抑制劑BrPa降低GLUT1的表達,緩解小鼠關節炎癥[7]。
2.1.2糖酵解途徑代謝酶 糖酵解過程中有幾種關鍵的限速酶參與了FLS侵襲性表型,第一種酶是HK2。與骨關節炎(osteoarthritis,OA)患者FLS相比,HK2在RA-FLS中表達增加,且脂多糖等炎癥介質能促進RA-FLS表達HK2,導致其遷移和增殖能力明顯增強,HK2基因沉默后,RA-FLS遷移和增殖能力降低。動物實驗表明,向K/BxN關節炎小鼠關節腔內注射HK2可增強FLS增殖和遷移能力,HK2敲除小鼠關節炎癥狀能得到明顯改善[8]。第二種酶是雙功能6-磷酸果糖-2-激酶/果糖-2,6-雙磷酸酶3(6-phosphofructo-2-kinase/fructose-2,6-biphosphatase3,PFKFB3)。RA患者滑膜組織中PFKFB3表達高于OA,PFKFB3小分子抑制劑PFK15可降低葡萄糖攝取和利用,抑制糖酵解途徑,顯著降低RA-FLS遷移、侵襲及產生炎癥介質的能力,減輕膠原誘導型關節炎小鼠關節炎的嚴重程度[9]。此外,RA患者CD4+T細胞PFKFB3缺乏活性,影響糖代謝途徑,ATP產生不足,容易導致細胞凋亡。第三種酶是丙酮酸激酶2(pyruvate kinase2,PKM2)。與OA相比,RA患者滑膜中PKM2的表達明顯增加,低氧環境下RA-FLS中PKM2表達升高,而通過PKM2抑制糖酵解途徑,可以逆轉RA-FLS侵襲、遷移及分泌功能[10]。
2.2 脂質代謝途徑
2.2.1脂質代謝途徑轉運體 磷脂酰膽堿是磷脂的主要成分之一,由膽堿合成。Seki等[11]發現膽堿類轉運體1(choline transporter-like 1,CTL1)(高親和力)和CTL2(低親和力)均在RA-FLS中高表達,與OA-FLS相比,RA-FLS膽堿攝取顯著增加。氟西汀等陽離子藥物可抑制膽堿攝取,顯著降低RA-FLS活力、增加細胞凋亡蛋白-3/7活性,可促進RA-FLS凋亡。此外,膽堿代謝與巨噬細胞介導的炎癥反應有關,M1巨噬細胞中CTL1表達增強,增加了膽堿攝取,進而促進磷脂酰膽堿生物合成,增加細胞因子分泌,促進巨噬細胞介導的炎癥免疫反應[12]。
血漿磷脂轉運蛋白(plasma phospholipid transfer protein,PLTP)是真核生物中普遍存在的轉運蛋白,PLTP可以通過其活性形式轉運脂質,也可以與ATP結合盒轉運蛋白A1(ATP-binding cassette transporter A1,ABCA1)等受體結合產生作用。研究表明,PLTP和ABCA1在RA-FLS中表達程度高,且RA-FLS中PLTP活性明顯高于OA,PLTP通過增加脂質轉運及與ABCA1結合激活STAT3 途徑,促進了RA-FLS增殖及分泌白介素8(interleukin-8,IL-8)、IL-6、MMP-3和血管內皮生長因子的能力,參與RA發病機制[13]。
2.2.2脂質代謝途徑代謝酶 磷脂酶A2(phospholipase A2,PLA2)可將花生四烯酸(arachidonic acid,AA)從細胞膜磷脂中游離出來,AA可通過環氧合酶(cyclooxygenase,COX)代謝成前列腺素,通過脂氧合酶(lipoxygenase,LOX)代謝成白三烯,這兩種代謝物均在體內充當炎性介質,介導RA患者關節炎癥的發生發展。PLA2、COX、LOX在RA患者滑膜中表達增高,提示AA代謝途徑的激活[14]。研究表明,LOX抑制劑能減少腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)誘導RA-FLS釋放炎癥因子,緩解關節炎小鼠的足爪腫脹[15];抑制PLA2可顯著降低TNF-α誘導FLS釋放AA、PGE2、IL-8和MMP-3[16];抑制COX的非甾體抗炎藥能有效地減輕RA患者臨床癥狀和體征,消除關節局部炎癥反應。
膽堿激酶α(choline kinase α,Chokα)是一種重要的磷脂酰膽堿合成酶,是細胞增殖的必需酶。研究表明,TNF-α和血小板源性生長因子可誘導RA-FLS中Chokα表達增加,使磷酸化膽堿水平升高。Chokα抑制劑MN58b緩解K/BxN關節炎小鼠炎癥反應程度,體外可抑制RA-FLS遷移并促進其凋亡[17]。磷脂酶D(phospholipase D,PLD)為磷脂酰膽堿的特異性切割酶,PLD1基因干擾和PLD1、PLD2小分子抑制劑均能顯著減少RA-FLS分泌IL-6、IL-8和趨化因子[18]。
2.3 氨基酸代謝途徑
2.3.1氨基酸代謝途徑轉運體 L型氨基酸轉運蛋白1(L-type amino acid transporter 1,LAT1)是普遍存在的L型氨基酸轉運蛋白家族成員之一,亮氨酸、異亮氨酸、色氨酸等均通過細胞LAT1轉運。研究發現LAT1在RA患者滑膜中表達增加,LAT1抑制劑BCH可降低mTOR及其下游靶基因4EBP1磷酸化、亮氨酸攝取和RA-FLS遷移[19]。Ozaki等[20]發現LAT1基因敲除的小鼠破骨細胞生成減少,體外研究發現LAT1通過mTOR復合體1通路調節破骨細胞形成,維持骨內環境穩定。
谷氨酰胺主要通過Na+依賴性丙氨酸-絲氨酸-半胱氨酸轉運載體2(alanine-serine-cysteine transporter 2,ASCT2)進行轉運,該轉運體參與T細胞與巨噬細胞活化,抑制ASCT2可以減少RA等自身免疫病中T細胞增殖和過度活化[21]。
2.3.2氨基酸代謝途徑代謝酶 色氨酸是機體必需氨基酸之一,RA等自身免疫病患者的血清和尿液中色氨酸水平降低、其分解代謝物水平升高[22]。色氨酸主要通過犬尿氨酸途徑進行分解代謝,吲哚胺-2,3-雙加氧酶1(indoleamine-2,3-dioxygenase 1,IDO1)、吲哚胺-2,3-雙加氧酶2(indoleamine-2, 3-dioxygenase2,IDO2)、色氨酸-2,3-雙加氧酶(tryptophan-2,3-dioxygenase 2,TDO2)是犬尿氨酸代謝途徑關鍵的限速酶。IDO1 抑制劑1-MT,可降低早期K/BxN關節炎小鼠B 細胞產生細胞因子和抗體的水平,從而減輕關節炎小鼠足爪腫脹程度。1-MT和MTX 聯合給藥,能更有效地緩解K/BxN小鼠關節炎癥[23]。IDO2是IDO1的同工酶,兩者具有43%的基因同源性,但IDO2酶解效率明顯低于IDO1。近年來研究發現,IDO2可能通過調控B 細胞和T細胞介導的免疫反應,也參與了自身免疫性關節炎的發生發展[22]。TDO2參與多種疾病發病,其在多種腫瘤組織中表達程度較高,且參與腫瘤的免疫反應。研究發現[24],神經膠質瘤患者中TDO2表達程度較高,而在TDO2表達高的部位中浸潤的CD8+T細胞水平大大降低;膠質瘤小鼠的T細胞TDO2表達增強,導致分泌的INF-γ水平明顯降低。此外,敲除TDO2基因的實驗性自身免疫性腦脊髓炎(experimental autoimmune encephalomyelitis,EAE)小鼠,緩解脊髓神經元的損傷,提示抑制TDO2可能成為多發性硬化癥等自身免疫病的新策略[25]。
Takahashi等[26]在2017年首次報道了谷氨酰胺在RA發病中的作用。谷氨酰胺酶1(glutaminase1,GLS1)是谷氨酰胺代謝途徑關鍵分子。研究發現,GLS1在RA-FLS中高表達,GLS1抑制劑化合物968可抑制RA-FLS生長,化合物968給藥后可減少自發性關節炎小鼠(SKG小鼠)關節炎評分及FLS數量,提示GLS 1在調節RA-FLS增殖中起重要作用。此外,BPTES能改善系統性紅斑狼瘡和EAE模型小鼠病理狀況,減少Th17細胞中低氧誘導因子1α表達,從而影響糖酵解途徑[27]。
3 小結
目前還沒有較好的治療RA的藥物,傳統藥物起效慢,副作用大,新型生物制劑也存在胃腸道反應、骨髓抑制等諸多不良反應[28]。對RA代謝內容的不斷認識與深入研究,不僅有助于理解該病的病理機制,更有望為RA臨床診斷提供新指標、為RA的治療提供新靶點和新方向。葡萄糖、脂質、氨基酸代謝途徑中轉運體及代謝酶等分子異常活化,導致RA-FLS代謝異常,參與RA發生發展。通過調節異常活化分子的活性,可改善能量代謝異常細胞的功能,從而達到緩解關節炎癥的效果。靶向FLS能量代謝異常關鍵分子,只針對功能異常細胞,使過度活化的細胞功能恢復至正常水平,在發揮治療作用的同時盡可能的降低不良反應,即達到“炎癥免疫反應軟調節”[29]目的,對未來RA藥物的開發具有重要意義。因此,全面、系統地闡述FLS代謝的變化,將為RA病理機制研究和藥物新靶點的發現提供重要理論依據和實驗依據。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
中國衛生(2016年3期)2016-11-12 13:23:26
中國衛生(2014年12期)2014-11-12 13:12:52
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
