Hsp90抑制劑的研究進展
2021-02-22 09:11:38張鐘元閻愛俠
中國醫藥生物技術 2021年1期
關鍵詞:結構
張鐘元,閻愛俠
·綜述·
Hsp90抑制劑的研究進展
張鐘元,閻愛俠
100029 北京化工大學化工資源有效利用國家重點實驗室/生命科學與技術學院
Hsp90 是最豐富的熱休克蛋白,它廣泛存在于真核以及原核生物中。Hsp90 的主要作用是幫助其客戶蛋白正確折疊和降解,這些客戶蛋白中有許多是在癌癥發生中起到重要作用的激酶和轉錄因子。還有許多客戶蛋白對于其他疾病的發展都是必不可少的,包括阿爾茨海默病和其他神經退行性疾病以及病毒和細菌感染。Hsp90 抑制劑通過與 Hsp90 調節位點結合引起 Hsp90 構象改變,并誘導底物蛋白降解,從而發揮抑制作用。雖然目前還沒有 Hsp90 抑制劑被批準上市,但是以 Hsp90 為靶點進行抑制劑的開發仍然具有重要意義,本文主要對其抑制劑的抗腫瘤活性研究進展進行綜述。
1 Hsp90 的結構特點
Hsp90 是高度保守的 ATP 依賴性熱應激蛋白,其表達是由應激相關轉錄因子熱休克因子 1(HSF1)誘導的,在應激耐受性和蛋白質折疊中起著非常重要的作用[1]。它是維持、激活或折疊特定客戶蛋白質的必需分子伴侶,這些客戶蛋白中有許多是信號通路中重要的蛋白質,包括 RAF 激酶蛋白、酪氨酸激酶受體 2(ErbB2)、Cdk4 和類固醇激素受體等[2]。
在人源 Hsp90 中已經鑒定出 4 種亞型:存在于胞質內的 Hsp90α 和 Hsp90β,存在于線粒體基質內的腫瘤壞死因子相關蛋白 1(TRAP1)以及內質網中的葡萄糖調節蛋白 94(Grp94)[3]。所有亞型都包含一個保守結構,包括 N 末端 ATP 酶結合域、中間域以及 C 末端結合域。由于 4 個亞型在細胞中的位置不同,所以它們各自結合的蛋白也不相同。
Hsp90 在細胞中以同源二聚體形式存在,每個單體由 3 個結構域組成:一個 N 末端結構域(NTD),一個中間域(MD)和一個C 末端結構域(CTD)[4]。NTD 是 Hsp90 的主要 ATP 酶結構域,與組氨酸激酶和拓撲異構酶具有高度的結構相似性。MD 在 ATP 水解以及客戶蛋白識別中起到重要作用,并且在 NTD 和 MD 中間由一段高電荷區域連接[5]。CTD 形成了 Hsp90 主要的二聚化界面,同時還包含 MEEVD 序列,該序列是對 TPR 域伴侶分子非常重要的相互作用位點[6]。
NTD 包含一個 ATP 結合位點,該位點負責 ATP 酶活性并提供伴侶蛋白循環所需的能量。NTD 包含 GHKL 超家族共有的一個“Bergerat fold”折疊區,該折疊區是一面由反平行的β 鏈,一面由單層α 螺旋組成的“雙層三明治”結構[7]。不同于蛋白激酶表現出的典型延伸構象,這個結合口袋的 ATP 以獨特的彎曲構象結合。ATP 結合口袋處于α 螺旋很深的裂隙中并一直延伸到疏水表面,天冬酰胺殘基結合的Mg2+離子通過溶劑介導作用將 ATP 所有的磷酸酯部分連接到蛋白質,腺嘌呤部分的 N6 位氨基與 Asp93 形成氫鍵[8]。在過去的數十年中,開發的所有 Hsp90 N 端抑制劑均是在此區域與 ATP 競爭性結合達到抑制 Hsp90 的作用。
Hsp90 的 MD 由大中間域以及小中間域兩部分組成,并且也是蛋白激酶 PKB/Akt 的結合位點[5]。由于 Hsp90 對 ATP 的親和力很低,因此 NTD 中的結合位點必須與 MD 接觸并且進行相互協同作用才能使 ATP 水解。
CTD 對于維持 Hsp90 同型二聚體的生物學活性構象非常重要,該結構域具有一個保守的 MEEVD 序列,該序列負責募集包含例如 Hsp70-Hsp90 組織蛋白在內的伴侶蛋白[9]。同時 CTD 包含第二個核苷酸結合位點,并且僅當 NTD 的 ATP 結合位點被占用時此位點才會被激活,此位點可變構調節 N 末端 ATP 酶的活性。NTD 對腺嘌呤具有特異性,而 CTD 的結合位點不僅可以結合嘌呤還可以結合嘧啶核苷酸[4]。
2 Hsp90 N 末端抑制劑
2.1 格爾德霉素及其衍生物
格爾德霉素(geldanamycin,GA)(圖 1)屬于苯醌安莎霉素,是鑒定出的第一個Hsp90 N 末端抑制劑,在過去幾十年中已成為基于天然產物藥物發現的原型[10]。Hsp90-GA復合物的晶體結構揭示了GA為 ATP 競爭性抑制劑,復合物的形成改變了 Hsp90 的構象,Hsp90 所需 ATP 酶的活性受到抑制,進而使得 Hsp90 不能與其客戶蛋白形成正確復合體,最終導致其客戶蛋白被蛋白酶體降解[11-12]。GA 的體外活性為Kd = 1.2 μmol/L,但在體內 Kd 值卻增加50 ~ 100 倍。由于體內活性不佳以及嚴重的肝毒性限制了其在臨床中的應用,針對 GA 的缺點進行改造,隨后產生了一系列 GA 衍生物。
17-烯丙基氨基-17-去甲氧基格爾德霉素(17-AAG)(圖 1)是 GA 第 17 位甲氧基被烯丙基氨基取代得到的衍生物,相比 GA,其生物活性有大幅提高(IC50= 31 nmol/L),并且毒性得到了改善[13]。17-AAG 已經完成 III 期臨床試驗[14],但是由于溶解度差以及脫靶毒性,其在治療晚期腫瘤方面的功效存在一定局限性。為了達到最佳治療效果,目前一般是將 17-AAG 與其他抗癌方案一起進行聯合給藥。17-AAG 還被發現可以通過削弱 T 細胞的功能從而減緩小鼠腦脊髓炎的發展;通過抑制 Hsp90 還破壞了 HIV 蛋白的表達;在體內靶向 Hsp90 還可以干擾諾如病毒的復制[14]。

圖 1 格爾德霉素及其衍生物的化學結構
17-二甲基氨基乙基氨基-17-去甲氧基格爾德霉素(17-DMAG)(圖 1)是 GA 第 17 位甲氧基被 N,N-二甲基乙胺取代得到的衍生物(IC50= 24 nmol/L),與 17-AAG 相比,由于可電離氨基的存在增加了水溶性,提高了口服生物利用度,并且具有更好的抗腫瘤活性[15]。17-DMAG 已經完成了 I 期臨床研究[16],根據結果顯示 II 期的推薦給藥劑量為每周 80 mg/m2。17-DMAG 可以增加 CD8 陽性 T 細胞,從而減輕蛋白尿癥;該藥物在處理大皰性表皮松懈的小鼠模型中有效,并且可以干擾包括 IFN-γ、TNF-α 和 IL-17 在內的炎癥因子的表達。
IPI-493(圖 1)是 GA 第 17 位甲氧基被氨基取代的衍生物,IPI-493 較 GA 相比生物活性大幅提高(EC50= 34 nmol/L),并且毒性得到改善[17]。IPI-493 對胃腸道間質瘤具有很強的抗腫瘤潛力,單一給藥時已經具有一定的抗腫瘤活性,當與伊馬替尼或舒尼替尼聯合使用時其治療效果會進一步增強。而且如果將 Hsp90 抑制劑與酪氨酸激酶抑制劑聯合使用可以潛在地克服腸道間質瘤患者對伊馬替尼或舒尼替尼的耐藥性問題[18]。
IPI-504(圖1)是 17-AAG 的水溶性氫醌鹽酸鹽衍生物(EC50= 64 nmol/L),具有比 17-AAG 更好的親和力以及水溶性,兩者通過氧化還原酶的作用在體內以氧化還原平衡存在[19]。IPI-504 曾經被認為是衍生自格爾德霉素類的最有希望的抑制劑,但是在多發性骨髓瘤、非小細胞肺癌、乳腺癌以及胃腸道間質瘤患者中進行的 I ~ III 期臨床試驗中或是未能達到預期的療效或是在治療組中觀察到較高的死亡率,臨床試驗均被終止。
2.2 間苯二酚衍生物
根赤殼菌素(radicicol,RD)是一種包含間苯二酚結構的大環內酯抗生素,最早是從真菌 Bonorden 分離得到的[20]。RD 的化學性質不穩定并且缺乏體內活性,但是進入臨床試驗中的許多抑制劑都包含間苯二酚核心結構,包括 NVP-AUY922、AT-13387 以及 ganetespib,這些小分子的結構見圖 2。雖然這些小分子都不是通過直接改造 RD 而得到的,但是它們將間苯二酚核心結構作為與 Hsp90 結合的關鍵基團而明顯類似于 RD 的作用效果。
NVP-AUY922 是基于 4,5-二芳基異噁唑骨架的 Hsp90小分子抑制劑,由英國癌癥研究中心的研究員采用基于結構的合理藥物設計方法合成,目前由諾華公司開發。在 Hsp90 熒光偏振競爭性結合實驗中,IC50達到了21 nmol/L 的高活性,體外實驗表明可以抑制多種人類癌細胞系的增殖[21]。在細胞水平上,NVP-AUY922 與阿霉素聯合用藥可以誘導 MCF-7 乳腺癌細胞株凋亡并下調血管內皮生長因子[22]。NVP-AUY922 主要用于患有復發或難治多發性骨髓瘤、非小細胞肺癌以及 HER2+和 Er+轉移性乳腺癌的患者,目前已經完成多項 I/II 期臨床試驗。NVP-AUY922 對于小鼠腦脊髓炎、HIV 蛋白的表達以及對諾如病毒的復制與 17-AAG 作用一致。

圖 2 間苯二酚衍生物的化學結構
AT-13387 是 Astex 公司科學家在進行片段篩選得到含有間苯二酚結構的目標化合物后,經過設計優化得到的小分子(Kd= 0.71 nmol/L)[23]。AT-13387 已經完成用于實體瘤患者的 I 期臨床試驗,用于胃腸道間質瘤經單獨或與伊馬替尼聯用給藥的 II 期臨床試驗,用于非小細胞肺癌單獨或與克唑替尼聯用給藥的 I、II 期臨床試驗,用于前列腺癌單獨或與乙酸阿比特龍聯用給藥的 I、II 期臨床試驗。
Ganetespib 是由 Synta 公司開發的含間苯二酚的三唑雜環化合物(IC50= 43 nmol/L),屬于第二代 Hsp90 抑制劑,對多種癌癥均具有抗腫瘤作用[24]。Ganetespib 用于 HER2 陽性轉移性乳腺癌患者的 I 期臨床試驗表明,與低濃度的紫杉醇以及曲妥珠單抗聯用的三聯體療法效果較好[25],顯示 II 期臨床的參考用藥量為 150 mg/m2;用于晚期食管胃癌患者的 II 期臨床試驗結果表現出可控的毒性,但是由于單獨給藥活性不足而被提前終止了。
2.3 嘌呤類衍生物
PU-3(圖 3)是 Chiosis 等[26]利用 Hsp90 與 ATP 酶結合具有的獨特折疊特征,采用基于結構的方法設計的第一個合成的 Hsp90 抑制劑(IC50= 0.55 μmol/L)。作為嘌呤支架類的原型,PU-3 已經通過多種策略進行結構優化,產生了一系列具有更優類藥性質的小分子 Hsp90 抑制劑。這些小分子具有一些共同特征:均包含一個連接于嘌呤或嘌呤樣核心的氨基基團和一個相距約 0.5 nm 的芳香環部分作為與 Hsp90 結合的關鍵基團[27]。已經進入臨床研究的小分子包括 PU-H71、BIIB021、BIIB028、MPC-3100 以及類似嘌呤的 Debio0932,這些小分子的結構見圖 3。
PU-H71 是 PU3 的衍生物(IC50= 90 nmol/L),對 Hsp90 的 ATP 結合域具有高溶解度和特異性[28]。據報道 PU-H71 在許多臨床前模型中均具有抗腫瘤作用,在三陰性乳腺癌細胞系中的 IC50為 65 ~ 140 nmol/L。針對實體瘤或淋巴瘤患者的兩項 I 期臨床試驗由于藥物供應中斷而提前結束了。該藥物自 2019 年以來正在積極招募患者進行晚期實體瘤、淋巴瘤和骨髓增生性疾病的 I 期臨床試驗評估。
BIIB021 是第一個基于口服、非格爾德霉素合成的 Hsp90 抑制劑(IC50= 310 nmol/L),在納摩爾濃度的臨床前模型中表現出抗腫瘤活性[29]。BIIB021 在嘌呤類別中具有結構特殊性,因為其芳基部分連接在嘌呤的 9 位,為了維持0.5 nm 的臨界距離而將氨基移至 2 位[30]。該藥物目前已經完成針對晚期實體瘤和淋巴瘤的 I 期臨床試驗和針對乳腺癌以及胃腸道間質瘤的 II 期臨床試驗。BIIB028 是第二代口服 Hsp90 抑制劑,是 BIIB021 的前藥[31]。其在效力和耐受性方面都比 BIIB021 有所改進,并且增加了靜脈給藥的治療窗口[32]。
MPC-3100 是由 Myrexis 公司開發的藥物(IC50=540 nmol/L),已經完成了針對難治性或復發性癌癥的 I 期臨床研究,然而不良的溶解性以及制劑問題限制了該藥物的進一步臨床開發[33]。為了解決溶解度和生物利用度較差的問題,又設計并優化得到了前藥 MPC-0767,該藥物克服了MPC-3100 暴露出的一些缺點,但是目前 MPC-0767 還在進行臨床前評估。

圖 3 嘌呤類衍生物的化學結構
Debio0932 是具有咪唑并吡啶結構的第二代口服 Hsp90 抑制劑(IC50= 100 nmol/L),由美國Curis 公司的科學家用碳置換了 N3 得到的嘌呤樣衍生物[34]。Debio0932 已經完成了針對晚期實體瘤或淋巴瘤的 I 期臨床研究,試驗結果顯示該藥物具有有限的臨床活性以及可控制的毒性,進一步可以發展成為非小細胞肺癌的輔助治療藥物[35]。
2.4 苯甲酰胺衍生物
苯甲酰胺化合物是另一類代表性的合成 Hsp90 抑制劑,苯甲酰胺基團模擬腺嘌呤,酰胺基團與 Asp93 和 Thr184 之間形成氫鍵。
SNX-5422(圖 4)是具有良好水溶性,可選擇性地與 ATP 口袋結合的口服 Hsp90 抑制劑(IC50= 32 nmol/L),它也是 SNX-2112 的前藥。SNX-5422 完成了針對晚期實體瘤和淋巴瘤患者的 I 期臨床試驗,盡管結果顯示具有良好的耐受性,也達到了預期的抑制效果,但是由于眼毒性以及潛在的不可逆性視網膜損傷的不良反應,終止了 SNX-5422 的開發[36]。
TAS-116(圖 4)是苯甲酰胺類衍生物,它是口服 Hsp90α/β 選擇性抑制劑,并不抑制 Hsp90 旁系同源物(Hsp90α,Ki= 34.7 nmol/L;Hsp90β,Ki= 21.3 nmol/L)[37]。TAS-116 已經完成了在實體瘤患者中的 I 期臨床研究,但是在臨床環境中普遍觀察到的不良事件是會不同程度導致視覺障礙[38]。
XL888(圖 4)是一種選擇性的 Hsp90 ATP 競爭性抑制劑。它可以抑制多種人類腫瘤細胞系的增殖,IC50值介于 0.1 ~ 45 nmol/L 之間,并導致包括 HER2、MET、突變型 BRAF 和突變型 EGFR 在內的客戶蛋白顯著降解[39]。目前正在針對大腸腺癌、轉移性胰腺腺癌、黑色素瘤以及皮膚癌招募患者準備進行 I 期臨床試驗。
2.5 其他類
Hsp990 是一種可口服生物利用的合成小分子,可通過 ATP 結合位點抑制 Hsp90(Hsp90α,IC50= 0.3 nmol/L;Hsp90β,IC50= 0.8 nmol/L),導致泛素蛋白酶體途徑驅動的客戶蛋白降解并抑制多種癌蛋白。它的抗腫瘤作用在不同惡性腫瘤的臨床前評估中得到證實,在針對晚期實體惡性腫瘤的 I 期臨床研究中表現出相對較好的耐受性,神經毒性是最相關的劑量限制性毒性[40]。
3 Hsp90 C 末端抑制劑
3.1 新生霉素及其衍生物
新生霉素和香豆素A1(圖 5)是從鏈霉菌種中分離得到的天然氨基香豆素類抗生素,也是第一個被報道的 C 末端 Hsp90 抑制劑[41]。盡管缺乏與抑制劑結合的 Hsp90 C 末端共晶體結構的報道,但是已經使用不同的技術來確認它們在 Hsp90 C 末端結合域的結合位點。使用固相結合測定方法、點突變分析方法、采用蛋白酶指紋圖譜以及生物信息學分析的綜合方法證實了新生霉素在該位點的結合[42]。
由于新生霉素對 Hsp90 的抑制活性較低(IC50約為700 nmol/L),因此通過構效關系研究設計優化得到了衍生物 A4[43]和 KU-32[44]。這些衍生物的結構可以模擬腺嘌呤和鳥嘌呤,并且提供氫鍵受體和供體以適應更高的特異性。
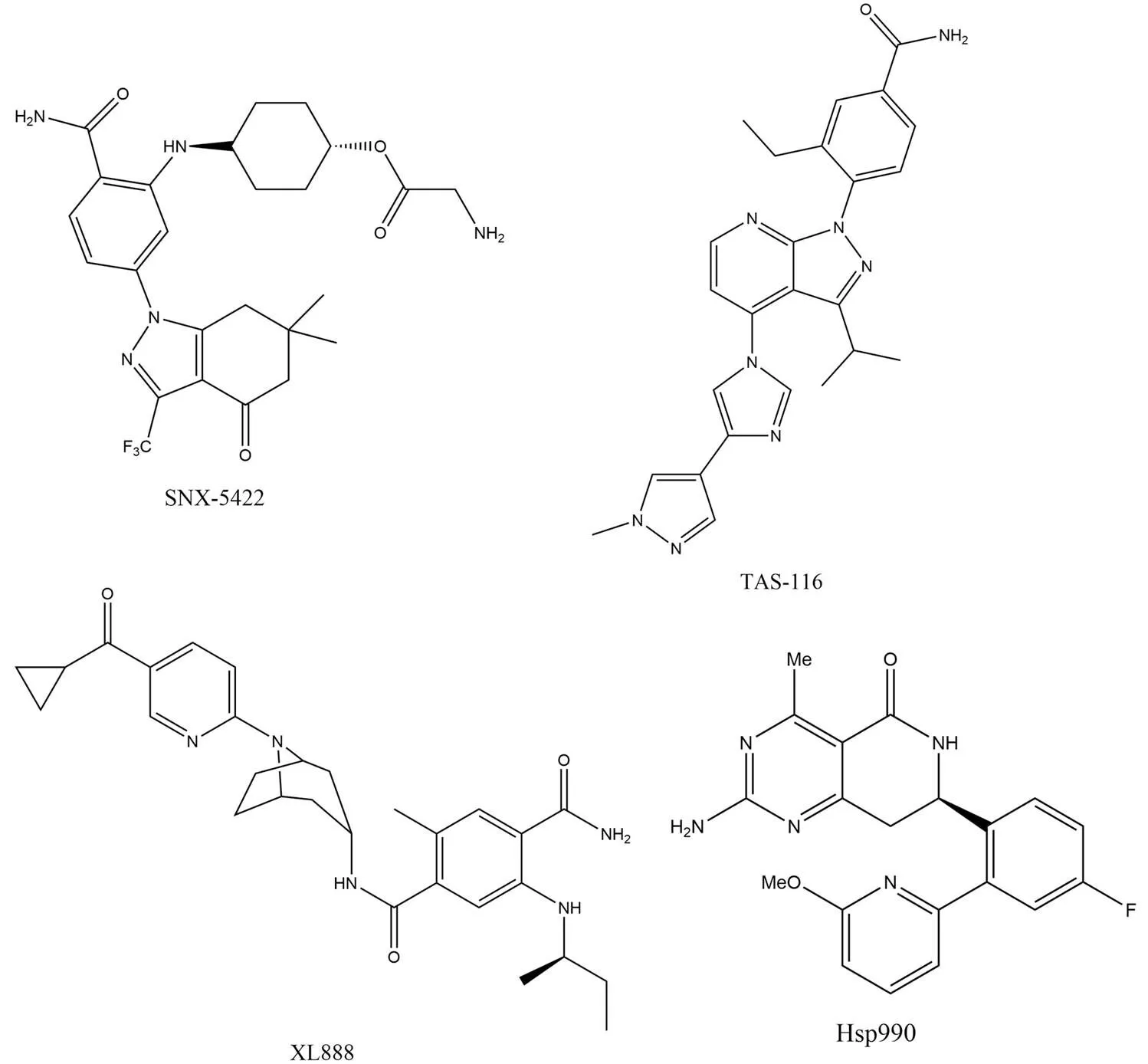
圖 4 苯甲酰胺衍生物以及 Hsp990 的化學結構
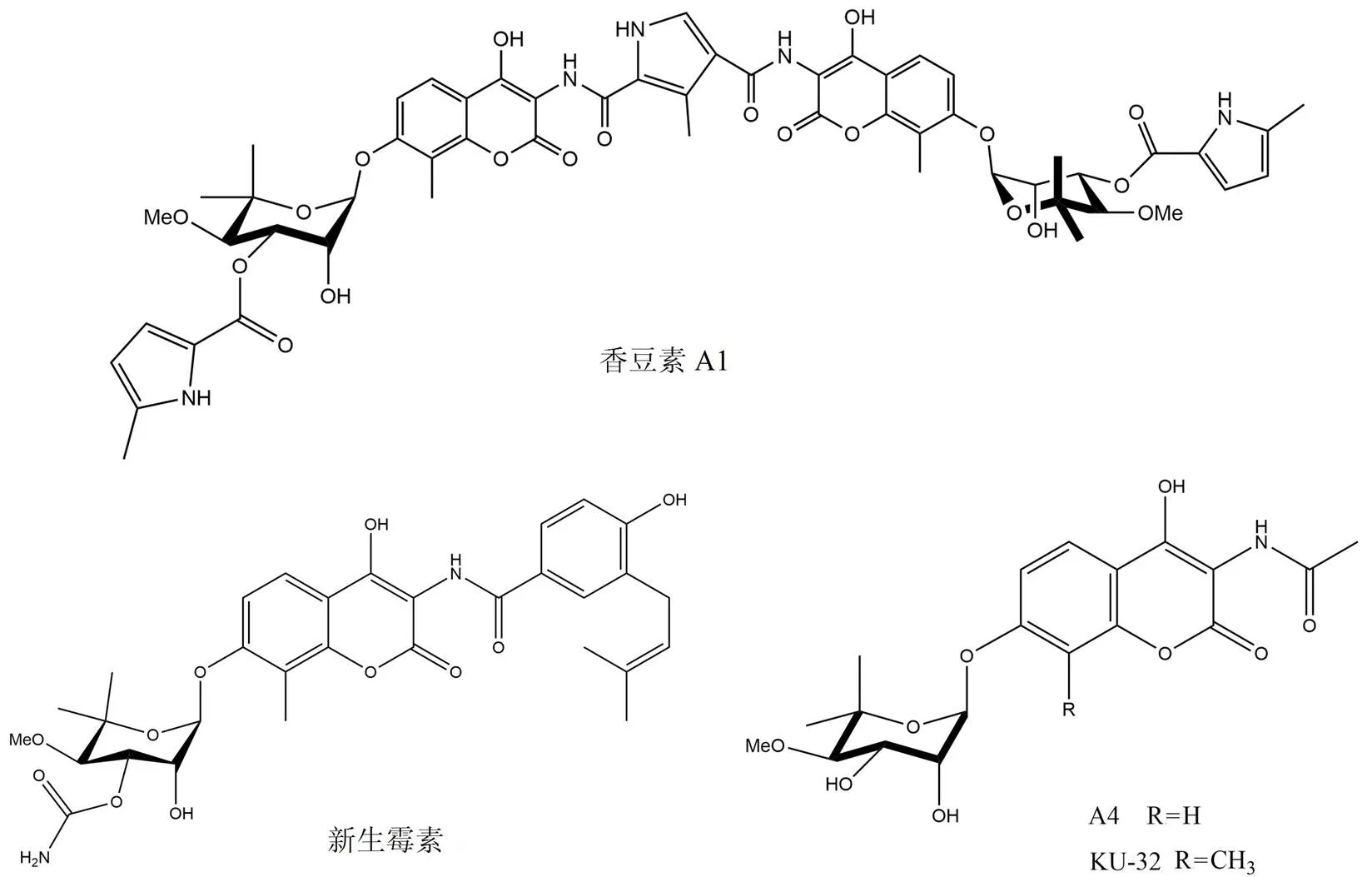
圖 5 新生霉素及其衍生物的化學結構
3.2 表沒食子兒茶素沒食子酸酯
表沒食子兒茶素沒食子酸酯(epigallocatechin gallate,EGCG)(圖 6)是一種來自綠茶茶樹的多酚[45],同新生霉素一樣也是與 C 端 ATP 結合位點結合的 Hsp90 抑制劑(IC50= 7.4 μmol/L)。EGCG 通過抑制 Hsp90 同型二聚體的功能降低了與癌癥相關的幾種 Hsp90 客戶蛋白的水平,如 ErbB2、Raf-1、phospho-AKT、pERK 以及 Bcl-2[46]。近年來已經開發了一系列 EGCG 的半合成和合成類似物,目的是克服其由多個酚羥基部分和其結構中代謝不穩定的酯引起的不良的類藥性質。這類化合物也在體外顯示出更強的Hsp90 抑制劑作用以及抗腫瘤功效[47]。
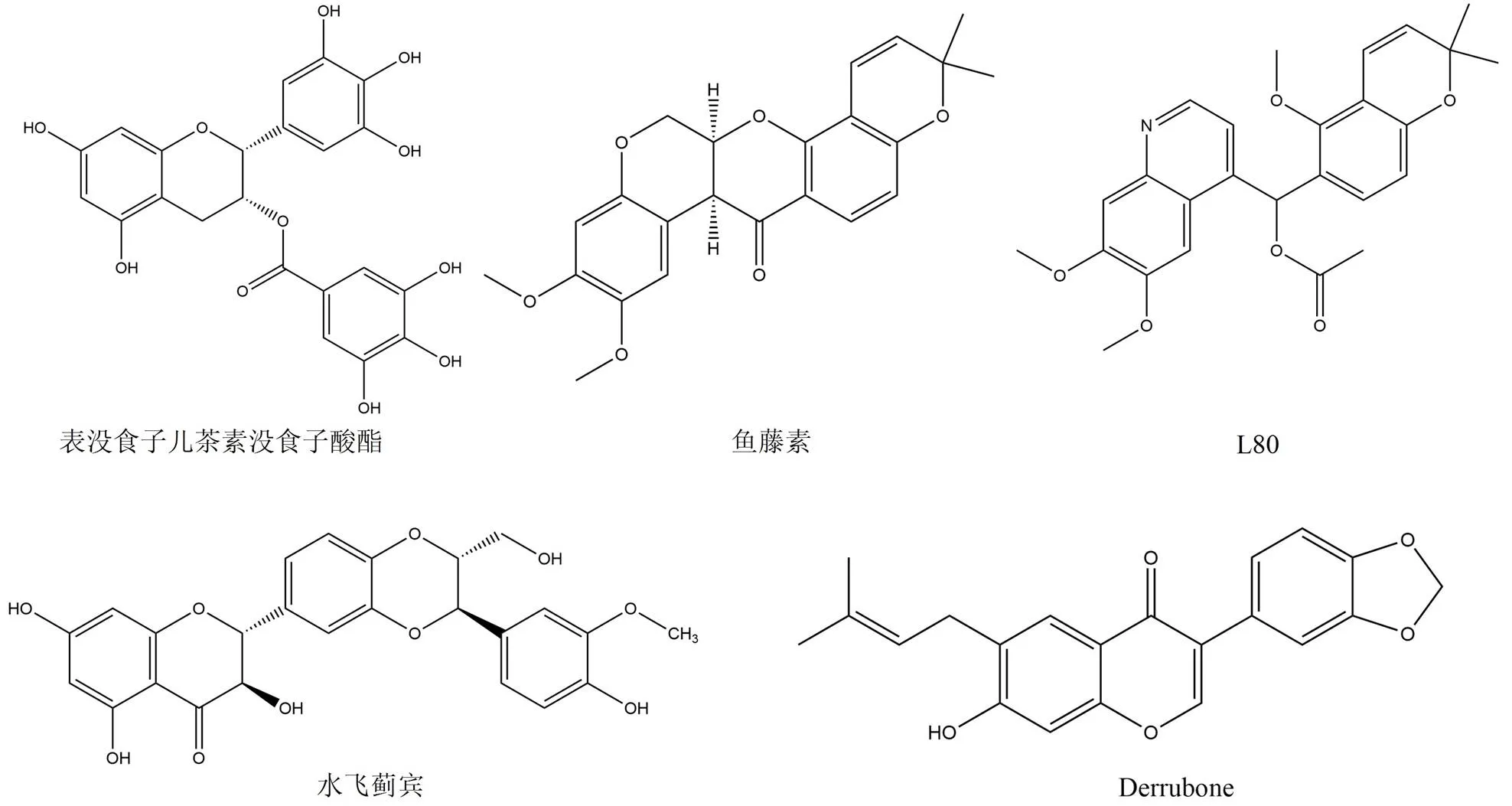
圖 6 EGCG 及黃酮類化合物的結構
3.3 黃酮以及類黃酮化合物
魚藤素(圖 6)是一種黃酮類化合物(IC50= 0.11 μmol/L),在各種癌細胞系以及動物實驗中均表現出優異的抗增殖和抗腫瘤血管生成特性,隨后的實驗證明其是通過抑制 Hsp90 C 端 ATP 結合口袋發揮作用的[48]。然而在大鼠的實驗中發現魚藤素會誘發帕金森病樣綜合征,為了克服這種不利因素對其進行了結構類似物設計和改造,產生了其結構類似物 L80。L80 可以誘導癌細胞凋亡,并導致 Hsp90 客戶蛋白降解以及磷酸化。通過實驗證明了 L80 的結合位點也是屬于 C 末端 ATP 結合區域,隨后的分子對接研究進一步證實了同型二聚體的結合模式,其與 K615、S677 以及 S677' 之間形成關鍵相互作用[49]。
水飛薊賓(圖 6)是從水飛薊果實中提取出的一種傳統黃酮類藥物,已被用于治療肝膽疾病很多年,但是水飛薊賓還顯示出對許多癌細胞系的抗癌活性。該藥物還被證明具有 Hsp90 C 端抑制活性,在導致依賴 Hsp90 客戶蛋白降解的同時不會改變 Hsp90 水平,IC50約為200 μmol/L[50]。由于新生霉素和水飛薊賓具有相似的生物學特性,因此開發了這兩種天然產物的嵌合衍生物,從而產生了具有更好的 Hsp90 抑制活性的化合物[51]。
Derrubone(圖 6)是通過高通量篩選分析發現的異戊烯基化的異黃酮類化合物(IC50= 9 μmol/L)。已有研究表明derrubone 及其合成類似物可以降解 Hsp90 客戶蛋白 HER2,并且抑制乳腺癌和結腸癌細胞系的體外增殖[52-53]。已經開發了一系列新生霉素和derrubone 的異黃酮嵌合體,但是這些嵌合物并沒有顯示出明顯的抗腫瘤作用[54]。
3.4 二氫吡啶
二氫吡啶已經被證實具有神經保護作用,在隨后的研究中顯示二氫吡啶 LA1011(圖 7)可以與 Hsp90 結合產生抑制作用。但是與其他大多數 Hsp90 抑制劑的作用不同,并沒有觀察到 Hsp90 ATP 酶的活性受到抑制。使用等溫滴定量熱法可以證明 LA1011 與 Hsp90 C 末端區域結合,但是目前并沒有充分的實驗證據表明 LA1011 結合在 C 末端 ATP 結合位點[55]。
3.5 順鉑和 LA-12
順鉑(圖 7)是目前常用的重金屬鉑類絡合物,由于分子中鉑的存在而廣泛用于治療各種癌癥及實體瘤[56]。順鉑的抗癌活性是由于其形成鏈內或鏈間 DNA 加合物的能力,同時也可以與各種蛋白質、磷脂以及 RNA 相互作用。順鉑可以選擇性地抑制 Hsp90 的某些功能,比如順鉑并不會誘導熱休克反應,經親和純化和蛋白質指紋圖譜進一步證實了順鉑與 Hsp90 C 末端區域結合。LA-12(圖 7)是順鉑優化后的衍生物,與順鉑相比具有更高的 Hsp90 親和力[57]。LA-12 可以誘導如突變體 p53、cyclin D1 和雌激素受體在內的一些 Hsp90 客戶蛋白的降解,并且對多種癌細胞系,包括對順鉑耐藥的癌細胞系也可以發揮抗腫瘤作用。
3.6 紫杉醇
紫杉醇(圖 7)是治療癌癥的常見藥物,其作用通過穩定微管和抑制有絲分裂實現。該藥物可以誘導轉錄因子和激酶激活,從而模仿細菌脂多糖(LPS)的作用。與典型的 Hsp90抑制劑相反,紫杉醇會表現出刺激性反應,介導巨噬細胞的激活并發揮 LPS 擬態效應。并且有研究證明 17-AAG 與紫杉醇誘導的細胞凋亡具有協同作用[58]。
4 干擾Hsp90 輔助伴侶結合的抑制劑
Hsp90 的功能依賴于其許多核苷酸影響的構象。Hsp90 和輔助分子伴侶 Hsp70、Hsp40、HOP、p23、Cdc37 與客戶蛋白等形成復合物保護客戶蛋白不被蛋白酶體所降解。考慮到不同的伴侶分子產生不同的作用機制,可以通過破壞Hsp90 與其輔助分子之間的蛋白-蛋白相互作用來實現選擇性的 Hsp90 抑制作用,從而降低毒性并提高選擇性。
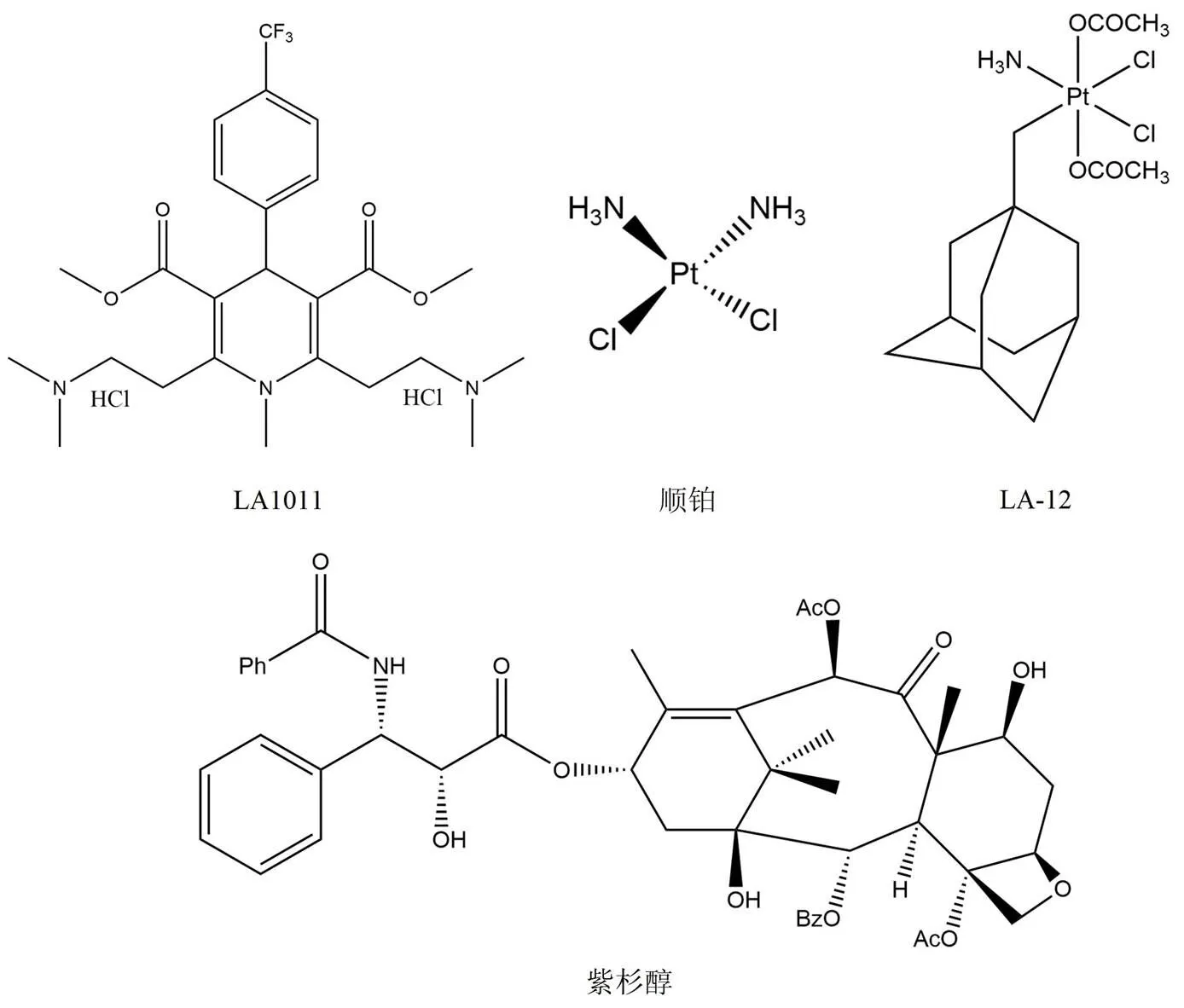
圖 7 LA1011、順鉑、LA-12 以及紫杉醇的化學結構
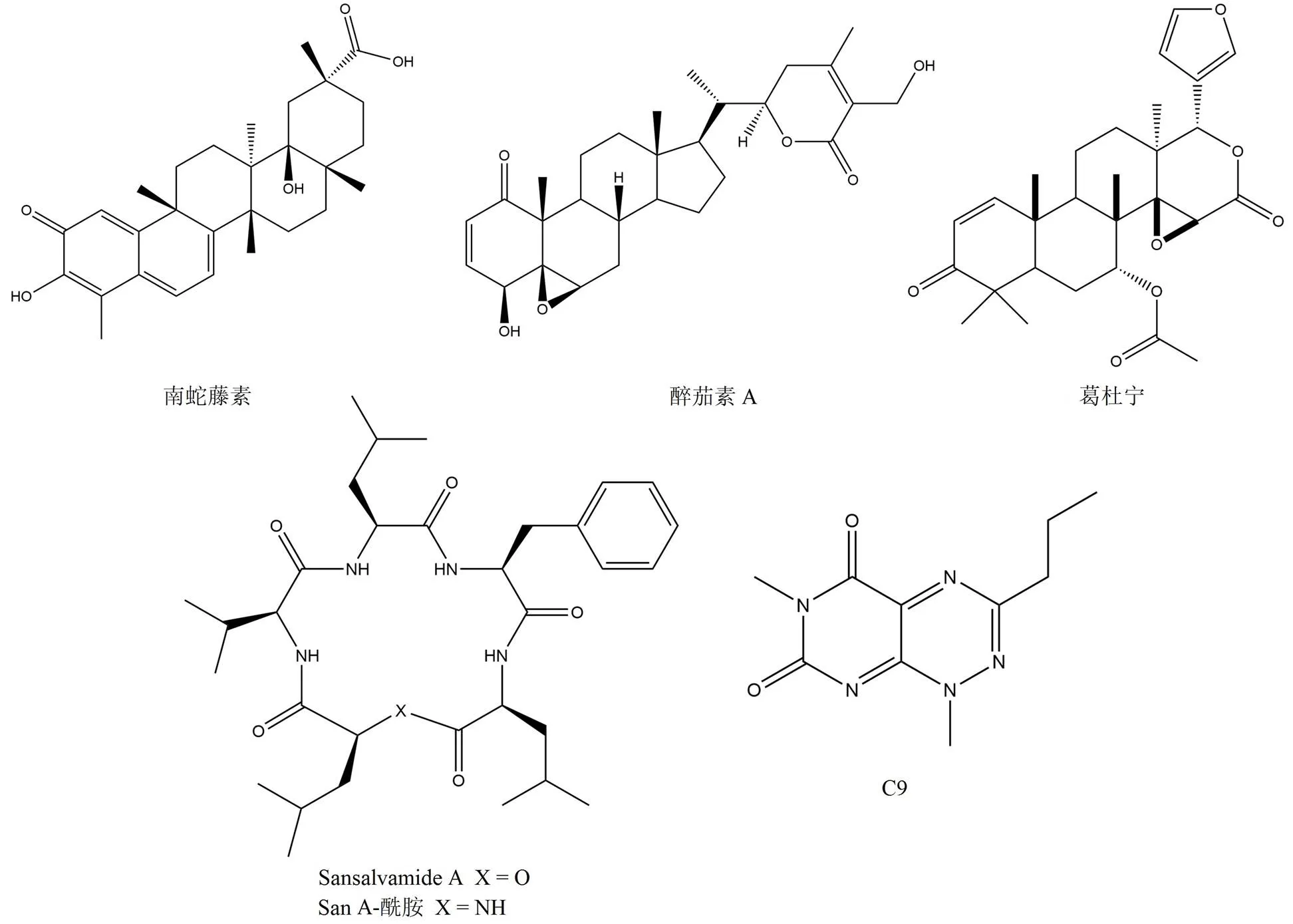
圖 8 干擾輔助伴侶結合的抑制劑化學結構
4.1 Hsp90-Cdc37 相互作用抑制劑
細胞分裂周期蛋白 Cdc37 可以幫助蛋白激酶結合到 Hsp90 上,這對于穩定 Hsp90 激酶復合物至關重要[59]。Cdc37 通過一個較大的相互作用表面與 Hsp90 相互作用,并且通過干擾 Hsp90 ATP 酶循環鏈過程中幾個關鍵點,從而阻止 ATP 水解[60]。因此,抑制 Hsp90-Cdc37 之間的結合也為其他抑制機制提供一個協同途徑。
南蛇藤素(圖 8)是從植物中提取的五環三萜類化合物[61],它可以激活熱休克因子 HSF-1 導致熱休克蛋白的表達(IC50= 0.3 μmol/L),具有抗增殖和神經保護的作用[62]。有研究表明,南蛇藤素通過共價結合 Cdc37 上的半胱氨酸殘基,從而阻止 Hsp90 及其協同分子 Cdc37 相互作用,進而導致客戶蛋白的選擇性降解,同時還可以防止胰蛋白酶對 C 末端區域的蛋白水解降解[63]。
類固醇內酯醉茄素 A(圖 8)具有抗增殖和抗腫瘤血管生成活性,在細胞水平上可以導致 Akt、CDK4 以及 GR 等客戶蛋白呈劑量依賴性降解。在免疫共沉淀實驗中該化合物顯示出阻斷 Hsp90 與 Cdc37 的相互作用(IC50= 1.24 μmol/L),由 Grover 等[64]進行的兩項分子對接研究提供了醉茄素 A在 Hsp90-Cdc37 界面可能的作用機制。
4.2 Hsp90-p23 相互作用抑制劑
小酸性蛋白 p23 通過 N 末端結構域和中間域與 Hsp90 相互作用,對于穩定類固醇激素受體是必需的[65]。葛杜寧(圖 8)是從印度楝樹中分離得到的三萜類化合物,在傳統印度醫學中已經用于抗瘧疾和其他傳染病。此外葛杜寧顯示出對包括前列腺癌、結腸癌和卵巢癌在內的各種癌細胞系的抗增殖活性,同時可以導致 Hsp90 客戶蛋白 Raf 和 HER2 降解(IC50= 8.84 μmol/L)[66]。在分子對接以及基因突變分析實驗中,該化合物與 p23 結合阻斷了 Hsp90 與其客戶蛋白結合并干擾小蛋白 p23 的基因調控[67]。
4.3 Hsp90-HOP 相互作用抑制劑
Hsp90 C 末端結構域的末尾包含一段高度保守的MEEVD 序列,該序列與 TRP 結構域形成關鍵的相互作用[68]。TRP 結構域存在于各種 Hsp90 共聚物中,其中之一就是 Hsp70/Hsp90 組織蛋白(HOP)[69]。
Sansalvamide A(San A)(圖 8)是從鐮刀菌屬海洋真菌中分離出的一種含有內酯部分的環狀五肽,具有中等的抗腫瘤活性(IC50= 45 μmol/L)[70]。隨后合成了 San A 的衍生物 San A-酰胺(圖 8)[71],發現其活性是天然產物的10 倍左右(IC50= 4.5 μmol/L)。在進行機制研究時發現其對 Hsp90 ATP 酶的活性沒有影響,破壞了 Hsp90 與 HOP 之間的相互作用。在過去的數十年中,開發了許多 San A-酰胺類似物,它們對包括胰腺癌、乳腺癌、前列腺癌和結腸癌在內的多種癌細胞系都表現出有效的細胞毒性[72]。
Yi 等[73]開發了一種高通量篩選測定法,該方法可以鑒定能夠抑制 Hsp90-TRP 相互作用的小分子。他們使用該方法測定了一組嘧啶并三嗪-二酮骨架,其中包括化合物 C9(圖 8)[74]。隨后驗證了體內活性,在治療人乳腺癌細胞 BT474 以及 SKBR3 時會降低客戶蛋白 HER2 的水平并誘導細胞凋亡。使用等溫滴定量熱法測量可以確定 C9 直接結合到了 TRP 域,C9 通過影響 Hsp90 依賴型蛋白折疊過程直接阻止了 Hsp90 與 HOP 的相互作用,進而對 Hsp90 產生了抑制作用[75]。
5 小結
數十年來,Hsp90 一直受到學術界和制藥行業的廣泛關注,N 末端抑制劑的開發已經取得重大進展,許多化合物已進行了臨床評估。然而這些抑制劑的一些諸如引起熱休克反應以及一系列毒性問題也日益凸顯。一些 C 末端抑制劑在臨床前研究中顯示出較好的結果,但是其治療效果尚未在患者中進行測試。現在隨著靶點作用機制更加清晰、計算模擬方法指導配體優化更加成熟以及尋求抑制 Hsp90 新途徑的開發,相信未來可以研發出用于臨床應用的新型 Hsp90 抑制劑。
[1] Maloney A, Workman P. HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert Opin Biol Ther, 2002, 2(1):3-24.
[2] Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst, 2000, 92(19):1564-1572.
[3] Blagg BSJ, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. Med Res Rev, 2006, 26(3):310-338.
[4] Harris SF, Shiau AK, Agard DA. The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure, 2004, 12(6):1087-1097.
[5] Jahn M, Rehn A, Pelz B, et al. The charged linker of the molecular chaperone Hsp90 modulates domain contacts and biological function. Proc Natl Acad Sci U S A, 2014, 111(50):17881-17886.
[6] Prodromou C, Roe SM, O'Brien R, et al. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell, 1997, 90(1):65-75.
[7] Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci, 2000, 25(1):24-28.
[8] Prodromou C, Roe SM, Piper PW, et al. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat Struct Biol, 1997, 4(6):477-482.
[9] Schmid AB, Lagleder S, Gr?wert MA, et al. The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J, 2012, 31(6):1506-1517.
[10] Whitesell L, Mimnaugh EG, De Costa B, et al. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A, 1994, 91(18): 8324-8328.
[11] Stebbins CE, Russo AA, Schneider C, et al. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell, 1997, 89(2):239-250.
[12] Roe SM, Prodromou C, O'Brien R, et al. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem, 1999, 42(2):260-266.
[13] Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol, 1998, 42(4):273-279.
[14] Chen F, Xie H, Bao H, et al. Combination of HSP90 and autophagy inhibitors promotes hepatocellular carcinoma apoptosis following incomplete thermal ablation. Mol Med Rep, 2020, 22(1):337-343.
[15] Hollingshead M, Alley M, Burger AM, et al. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother Pharmacol, 2005, 56(2):115-125.
[16] Pacey S, Wilson RH, Walton M, et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res, 2011, 17(6):1561-1570.
[17] Tian ZQ, Liu Y, Zhang D, et al. Synthesis and biological activities of novel 17-aminogeldanamycin derivatives. Bioorg Med Chem, 2004, 12(20):5317-5329.
[18] Floris G, Sciot R, Wozniak A, et al. The novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin Cancer Res, 2011, 17(17):5604-5614.
[19] Sydor JR, Normant E, Pien CS, et al. Development of 17-allylamino- 17-demethoxygeldanamycin hydroquinone hydrochloride (IPI-504), an anti-cancer agent directed against Hsp90. Proc Natl Acad Sci U S A, 2006, 103(46):17408-17413.
[20] Delmotte P, Delmotte-Plaque J. A new antifungal substance of fungal origin. Nature, 1953, 171(4347):344.
[21] Brough PA, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem, 2008, 51(2):196-218.
[22] Mohammadian M, Feizollahzadeh S, Mahmoudi R, et al. Hsp90 inhibitor; NVP-AUY922 in combination with doxorubicin induces apoptosis and downregulates VEGF in MCF-7 breast cancer cell line. Asian Pac J Cancer Prev, 2020, 21(6):1773-1778.
[23] Woodhead AJ, Angove H, Carr MG, et al. Discovery of (2,4-dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydrois oindol-2-yl]methanone (AT13387), a novel inhibitor of the molecular chaperone Hsp90 by fragment based drug design. J Med Chem, 2010, 53(16):5956-5969.
[24] Ying W, Du Z, Sun L, et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol Cancer Ther, 2012, 11(2):475- 484.
[25] Jhaveri K, Wang R, Teplinsky E, et al. A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res, 2017, 19(1):89-97.
[26] Chiosis G, Timaul MN, Lucas B, et al. A small molecule designed to bind to the adenine nucleotide pocket of Hsp90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol, 2001, 8(3):289-299.
[27] Taldone T, Chiosis G. Purine-scaffold Hsp90 inhibitors. Curr Top Med Chem, 2009, 9(15):1436-1446.
[28] Caldas-Lopes E, Cerchietti L, Ahn JH, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A, 2009, 106(20):8368-8373.
[29] Lundgren K, Zhang H, Brekken J, et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol Cancer Ther, 2009, 8(4):921-929.
[30] Kasibhatla SR, Hong K, Biamonte MA, et al. Rationally designed high-affinity 2-amino-6-halopurine heat shock protein 90 inhibitors that exhibit potent antitumor activity. J Med Chem, 2007, 50(12): 2767-2778.
[31] Lundgren K, Biamonte MA. The discovery of BIIB021 and BIIB028. //Machajewski TD, Gao Z. Inhibitors of molecular chaperones as therapeutic agents. RSC Publishing, 2013:158-179.
[32] Hong D, Said R, Falchook G, et al. Phase I study of BIIB028, a selective heat shock protein 90 inhibitor, in patients with refractory metastatic or locally advanced solid tumors. Clin Cancer Res, 2013, 19(17):4824-4831.
[33] Kim SH, Tangallapally R, Kim IC, et al. Discovery of an L-alanine ester prodrug of the Hsp90 inhibitor, MPC-3100. Bioorg Med Chem Lett, 2015, 25(22):5254-5257.
[34] Bao R, Lai CJ, Qu H, et al. CUDC-305, a novel synthetic HSP90 inhibitor with unique pharmacologic properties for cancer therapy. Clin Cancer Res, 2009, 15(12):4046-4057.
[35] Stenderup K, Rosada C, Gavillet B, et al. Debio 0932, a new oral Hsp90 inhibitor, alleviates psoriasis in a xenograft transplantation model. Acta Derm Venereol, 2014, 94(6):672-676.
[36] Rajan A, Kelly RJ, Trepel JB, et al. A phase i study of PF-04929113 (SNX-5422), an orally bioavailable heat shock protein 90 inhibitor, in patients with refractory solid tumor malignancies and lymphomas. Clin Cancer Res, 2011, 17(21):6831-6839.
[37] Ohkubo S, Kodama Y, Muraoka H, et al. TAS-116, a highly selective inhibitor of heat shock protein 90α and β, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Cancer Ther, 2015, 14(1):14-22.
[38] Doi T, Kurokawa Y, Sawaki A, et al. Efficacy and safety of TAS-116, an oral inhibitor of heat shock protein 90, in patients with metastatic or unresectable gastrointestinal stromal tumour refractory to imatinib, sunitinib and regorafenib: a phase II, single-arm trial. Eur J Cancer, 2019, 121:29-39.
[39] Bussenius J, Blazey CM, Aay N, et al. Discovery of XL888: A novel tropane-derived small molecule inhibitor of HSP90. Bioorg Med Chem Lett, 2012, 22(17):5396-5404.
[40] Spreafico A, Delord JP, De Mattos-Arruda L, et al. A first-in-human phase I, dose-escalation, multicentre study of HSP990 administered orally in adult patients with advanced solid malignancies. Br J Cancer, 2015, 112(4):650-659.
[41] Marcu MG, Chadli A, Bouhouche I, et al. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem, 2000, 275(47):37181-37186.
[42] Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst, 2000, 92(3):242-248.
[43] Yu XM, Shen G, Neckers L, et al. Hsp90 inhibitors identified from a library of novobiocin analogues. J Am Chem Soc, 2005, 127(37): 12778-12779.
[44] Kusuma BR, Zhang L, Sundstrom T, et al. Synthesis and evaluation of novologues as C-terminal Hsp90 inhibitors with cytoprotective activity against sensory neuron glucotoxicity. J Med Chem, 2012, 55(12):5797-5812.
[45] Kada T, Kaneko K, Matsuzaki S, et al. Detection and chemical identification of natural bio-antimutagens. A case of the green tea factor. Mutat Res, 1985, 150(1-2):127-132.
[46] Yin Z, Henry EC, Gasiewicz TA. (-)-Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry, 2009, 48(2):336-345.
[47] Khandelwal A, Hall JA, Blagg BSJ. Synthesis and structure-activity relationships of EGCG analogues, a recently identified Hsp90 inhibitor. J Org Chem, 2013, 78(16):7859-7884.
[48] Sgobba M, Degliesposti G, Ferrari AM, et al. Structural models and binding site prediction of the C-terminal domain of human Hsp90: A new target for anticancer drugs. Chem Biol Drug Des, 2008, 71(5): 420-433.
[49] Lee SC, Min HY, Choi H, et al. Synthesis and evaluation of a novel deguelin derivative, L80, which disrupts ATP binding to the C-terminal domain of heat shock protein 90. Mol Pharmacol, 2015, 88(2):245-255.
[50] Zhao H, Brandt GE, Galam L, et al. Identification and initial SAR of silybin: an Hsp90 inhibitor. Bioorg Med Chem Lett, 2011, 21(9): 2659-2664.
[51] Zhao H, Yan B, Peterson LB, et al. 3-Arylcoumarin derivatives manifest anti-proliferative activity through Hsp90 inhibition. ACS Med Chem Lett, 2012, 3(4):327-331.
[52] Hastings JM, Hadden MK, Blagg BSJ. Synthesis and evaluation of derrubone and select analogues. J Org Chem, 2008, 73(2):369-373.
[53] Hadden MK, Galam L, Gestwicki JE, et al. Derrubone, an inhibitor of the Hsp90 protein folding machinery. J Nat Prod, 2007, 70(12):2014- 2018.
[54] Mays JR, Hill SA, Moyers JT, et al. The synthesis and evaluation of flavone and isoflavone chimeras of novobiocin and derrubone. Bioorg Med Chem, 2010, 18(1):249-266.
[55] Kasza á, Hunya á, Frank Z, et al. Dihydropyridine derivatives modulate heat shock responses and have a neuroprotective effect in a transgenic mouse model of Alzheimer's disease. J Alzheimers Dis, 2016, 53(2):557-571.
[56] Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci, 2000, 57(8-9):1229-1235.
[57] Kvardova V, Hrstka R, Walerych D, et al. The new platinum (IV) derivative LA-12 shows stronger inhibitory effect on Hsp90 function compared to cisplatin. Mol Cancer, 2010, 9:147.
[58] Donnelly A, Blagg BSJ. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem, 2008, 15(26):2702-2717.
[59] Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell, 2012, 150(5):987-1001.
[60] Roe SM, Ali MMU, Meyer P, et al. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50cdc37. Cell, 2004, 116(1):87-98.
[61] Westerheide SD, Bosman JD, Mbadugha BNA, et al. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem, 2004, 279(53):56053-56060.
[62] Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell, 2006, 10(4):321-330.
[63] Zhang T, Li Y, Yu Y, et al. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem, 2009, 284(51):35381- 35389.
[64] Grover A, Shandilya A, Agrawal V, et al. Hsp90/Cdc37 chaperone/co-chaperone complex, a novel junction anticancer target elucidated by the mode of action of herbal drug Withaferin A. BMC Bioinformatics, 2011, 12 Suppl 1(Suppl 1):S30.
[65] Ali MMU, Roe SM, Vaughan CK, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature, 2006, 440(7087):1013-1017.
[66] Brandt GE, Schmidt MD, Prisinzano TE, et al. Gedunin, a novel hsp90 inhibitor: semisynthesis of derivatives and preliminary structure-activity relationships. J Med Chem, 2008, 51(20):6495-6502.
[67] Patwardhan CA, Fauq A, Peterson LB, et al. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis.J Biol Chem, 2013, 288(10):7313-7325.
[68] Horibe T, Kohno M, Haramoto M, et al. Designed hybrid TPR peptide targeting Hsp90 as a novel anticancer agent. J Transl Med, 2011, 9:8.
[69] Ciglia E, Vergin J, Reimann S, et al. Resolving hot spots in the C-terminal dimerization domain that determine the stability of the molecular chaperone Hsp90. PLoS One, 2014, 9(4):e96031.
[70] Carroll CL, Johnston JVC, Kekec A, et al. Synthesis and cytotoxicity of novel sansalvamide A derivatives. Org Lett, 2005, 7(16):3481- 3484.
[71] Alexander LD, Partridge JR, Agard DA, et al. A small molecule that preferentially binds the closed conformation of Hsp90. Bioorg Med Chem Lett, 2011, 21(23):7068-7071.
[72] Kunicki JB, Petersen MN, Alexander LD, et al. Synthesis and evaluation of biotinylated sansalvamide A analogs and their modulation of Hsp90. Bioorg Med Chem Lett, 2011, 21(16):4716- 4719.
[73] Yi F, Zhu P, Southall N, et al. An alpha screen-based high-throughput screen to identify inhibitors of Hsp90-cochaperone interaction. J Biomol Screen, 2009, 14(3):273-281.
[74] Yi F, Regan L. A novel class of small molecule inhibitors of Hsp90. ACS Chem Biol, 2008, 3(10):645-654.
[75] Pimienta G, Herbert KM, Regan L. A compound that inhibits the HOP-Hsp90 complex formation and has unique killing effects in breast cancer cell lines. Mol Pharm, 2011, 8(6):2252-2261.
國家自然科學基金(21675010)
閻愛俠,Email:yanax@mail.buct.edu.cn
2020-09-01
10.3969/j.issn.1673-713X.2021.01.008
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
