水產品中有機危害物殘留檢測技術挑戰及流程展望
2021-02-25 02:40:28高磊王鐘強覃東立陳中祥王鵬
水產學雜志 2021年6期
關鍵詞:檢測
高磊,王鐘強,覃東立,陳中祥,王鵬
(1.中國水產科學研究院黑龍江水產研究所,黑龍江 哈爾濱 150070;2.農業農村部漁業環境及水產品質量監督檢驗測試中心(哈爾濱),黑龍江 哈爾濱 150070;3.農業農村部水產品質量安全控制重點實驗室,北京 100141;4.河北省水產技術推廣總站,河北 石家莊 050021)
關鍵字:水產品質量安全;檢測技術;有機危害物
1 水產品中有機危害物殘留檢測中的挑戰
1.1 水產品中有機危害物殘留現狀
農業農村部辦公廳于2018 年發布《鄉村振興科技支撐行動實施方案》,倡導提質增效模式[1],目前應提質增效為導向,加快推進水產養殖業綠色發展。民以食為天,食以安為先,確保老百姓“舌尖上的安全”,關系到人民健康生活。目前農業農村部等相關部門相繼推出了多種檢測制度。根據其公開數據顯示,2013 年以來,隨著國家養殖水產品和苗種產地抽查等任務的開展,截至2019 年均沒有發生重大水產品質量安全事件,連續6 年合格率保持在99%以上[2,3]。
1.2 發現問題及存在風險
雖然所監測的水產品中常用藥物(包括但不限于硝基呋喃類代謝物、孔雀石綠類、磺胺類、喹諾酮類和氯霉素類等)已經得到了很好的控制。但仍然存在著如檢測周期長、非靶向有害物污染、假陰性、假陽性及標準規定滯后性等挑戰。
1.2.1 檢測周期長
參考農業部783 號公告-1-2006 標準以水產品中硝基呋喃類代謝物的檢測為例,需要在檢測前經過樣品采集、制備、前處理及儀器分析檢測過程[樣品制備(魚體→去皮→剔肉→攪碎→四分→勻漿→分裝)→冷鏈帶回實驗室→待測物提取富集凈化(加入內標→加入衍生化試劑→加入鹽酸→震蕩16 h→加入緩沖鹽→加入萃取劑→渦旋→離心→去上清液→再次加入萃取劑→渦旋→離心→取上清液→合并提取液→氮氣吹干→重新溶解→過濾→裝進樣瓶)→儀器分析檢測],其復雜的前處理流程往往需要2 d 以上。而其中的儀器分析檢測部分往往用到液相色譜串聯質譜等高精度的大型檢測儀器,一般需要恒溫、恒濕、無振動的工作環境,因而必須在實驗室進行樣品分析。待檢測完畢再出具檢測報告往往需要等到第三天之后,不合格的水產品有流入市場的風險,存在檢測周期長的缺陷。
1.2.2 非靶向有害物污染
目前的檢測標準往往是靶向檢測,而針對非靶向有機污染物卻無能為力。隨著稻魚互作、稻蟹互作等養殖方式在全國推廣,種植用田中所用的農藥可能通過雨水沖刷、地表徑流等作用滲透到池塘中,引起池塘中除草劑的殘留污染。而這些非靶向監測物質很可能影響水產品質量安全。2017 年,喬丹[4]對山東沿海13 個縣區貝類養殖基地中16 種除草劑(含酰胺類除草劑:乙草胺、異丙甲草胺)進行篩查,發現檢出率高達69.7%,而國外也在環境中發現了大量酰胺類除草劑的殘留[5]。本文前期調查研究中也發現黑龍江漁業水域及水產品中有丁草胺殘留。酰胺類除草劑(甲草胺、丁草胺等)被我國分類為高毒級的農藥,美國環境保護局也將甲草胺、乙草胺和丁草胺等定義為B-2 類致癌物,異丙甲草胺定位C 類致癌物[6]。其中甲草胺和乙草胺能顯著增強人類淋巴細胞姊妹染色體的交換頻率,降低人類精子的存活率和運動性[7]。丁草胺具有致突變性[8],會提高染色體畸變概率[9]。酰胺類除草劑在水生生物中的毒性更強,比哺乳動物毒性高500~10 000 倍[10]。高濃度丁草胺可誘發黃鱔(Monopterus albus)染色體數目畸變,染色體單體發生裂隙、斷裂[11]。乙草胺、丁草胺和異丙甲草胺在大鼠體內經細胞色素P450 酶系、芳基酰胺酶等一系列催化作用下會形成強致癌作用的二烷基醌亞胺[12]。而丁草胺這種除草劑往往并不在水產品檢測的靶向列表中,可能會面臨非靶向有機危害物的污染問題。
1.2.3 氨基脲的假陽性及檢測的假陰性
根據王建的研究總結[13],孟加拉國蝦的外殼存在著高含量的氨基脲(SEM),它在小龍蝦(Procambarus clarkii)肉中呈游離狀態,含量在0.4~12 μg/kg,其是否存在與添加呋喃西林沒有任何的關系。在食品工業中用來增稠的卡拉膠很可能是SEM的一種來源,因而其認為SEM 作為唯一的標志物來檢測呋喃西林已經不現實。因此,現行的檢測標準有可能會因為內源性的問題誤判為陽性,存在假陽性檢測風險。
目前如液相色譜紫外,液相色譜熒光、氣相色譜電子捕獲和氣相色譜火焰離子化等檢測器的特異性不強,其定性往往主要依靠保留時間,當基質干擾較大時,往往會造成目標出峰時間周圍有較大響應信號的共流出物,造成檢測結果的假陰性。而目前較為先進的定性手段多采用LC-MS/MS 確證,而質譜法本身的儀器特點,當涉及到高濃度鹽、非離子型表面活性劑等因素的干擾,也會影響其在電噴霧電離源的電離,引起檢測結果的不確定性,甚至引起假陰性[14],也會存在一小部分假陰性檢測風險。
1.2.4 部分檢測標準或規定的滯后性
采用的部分檢測標準往往還較為滯后,例如孔雀石綠采用《GB-T-19857-2005 水產品中孔雀石綠和結晶紫殘留量的測定》檢測方案,硝基呋喃類代謝物采用《農業部783 號公告-1-2006 水產品中硝基呋喃類代謝物殘留量的測定液相色譜-串聯質譜法》檢測方案,磺胺及喹諾酮類抗生素采用《農業部1077 號公告-1-2008 水產品中17 種磺胺類及15 種喹諾酮類藥物殘留量的測定液相色譜-串聯質譜法》檢測方案,氯霉素,氯霉素、甲砜霉素和氟苯尼考《GBT 20756-2006 可食動物肌肉、肝臟和水產品中氯霉素、甲砜霉素和氟苯尼考殘留量的測定液相色譜-串聯質譜法》檢測方案。其標準大多為2005—2008 年的標準,距今已有十余年,因此標準具有一定的滯后性。
目前檢驗檢測增殖放流經濟水產苗種中的氯霉素仍按照《農業部辦公廳關于開展增殖放流經濟水產苗種質量安全檢驗的通知》(農辦漁[2009]52號)[15]規定,采用酶聯免疫試劑盒ELISA+氣相色譜儀確證或氣相色譜質譜法規定的方法檢驗水產苗種藥殘。而《SC/T 3018-2004 水產品中氯霉素殘留量的測定氣相色譜法》方法涉及到提取→脫脂凈化→C18 柱凈化→衍生化(標準溶液亦需衍生化)→儀器分析測定,其相比于《GB/T 20756-2006 可食動物肌肉、肝臟和水產品中氯霉素、甲砜霉素和氟苯尼考殘留量的測定液相色譜-串聯質譜法》液相色譜串聯質譜法(提取→凈化→儀器分析測定)操作復雜。LC-MS/MS 采用了多反應監測模式會使得氯霉素的定性更加準確,抗基質能力更強,得到更低的檢出限(LC-MS/MS:0.1 μg/kg;GC-ECD:0.3 μg/kg)。據此,建議可對氯霉素的LC-MS/MS 方法進行驗證,如方法具備可行性,可使有條件的檢驗檢測機構選優先選用液相色譜串聯質譜測定氯霉素,可增加其作為常規的檢測方法,以提高檢測效率和定性準確性,獲得更好的檢出限。
以往傳統的檢測技術因前處理或分析過程有檢測周期長的問題;所常用的標準檢測手段往往是靶向檢測,無法針對非靶向物質進行篩查,違法者可通過避開靶向檢測物以達到檢測結果合格的目的,存在著逃避靶向檢測的風險;而檢測技術的不完善也制約著檢測結果,有可能出現檢測結果的假陽性、假陰性、標準滯后性等問題。因此,未來可通過改變當前的檢測流程來完善水產品質量安全體系。
2 未來水產品有機危害物檢測技術流程的展望
未來水產品檢測技術流程可采用“先撒網篩查,后準確定性”的檢測方針,具體如圖1 所示。
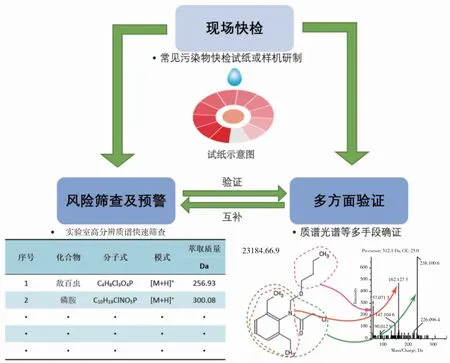
圖1 水產品檢測流程展望Fig.1 Process of fishery product detection technology
2.1 現場快檢
為解決傳統檢測周期長的問題,可應用不同的機理設計出對常規靶向待測物的快檢試劑盒,優先應用于活體現場快速檢測,在水產品流入市場前即可獲得準確結果。目前較為成熟的技術包括化學比色法[16]、酶聯免疫法[17,18]、膠體金免疫層析法[19,20]、量子點熒光免疫分析法[21,22]及表面增強拉曼光譜[23]等。開發出更為快速、高靈敏度的前處理及準確定性定量的現場快檢技術,在短時間內準確定性定量,不需要復雜的人和機器,使漁民及監管機構買得起、用的起、用的懂。解決傳統采樣→制樣→回實驗室→前處理→儀器分析檢測過程中檢測周期長帶來的風險,在水產品流通到市場前得到有效的風險排查。食品中部分快檢技術已經得到了應用[24,25],在監管中發揮重要作用[26,27]。除了要從源頭上控制水產品質量安全,還需要可靠的現場快檢技術作為支撐以保障人們的食品安全。
2.2 風險篩查及預警
為控制非靶向有害物、假陰性及假陽性的風險,可采取風險篩查及風險預警的措施。
2.2.1 風險篩查
針對目前的有害物質制定篩查表格(下至幾百種,上至上萬種),采用高分辨質譜對表格中有機危害物進行高通量篩查,不僅可篩查待測的水產品,亦可同時篩查漁業環境樣品(如水環境,底泥環境等)和漁業投入品等。選擇不同性質的物質作為質控樣品,驗證方法準確性,通過保留時間、精確分子量、同位素豐度比、二級譜圖等多方面信息提早發現風險物質。篩查的目的為盡可能的保留全部信息,其前處理過程較靶向檢測簡單,高分辨質譜數據具有可追溯性,可在多年后分析當年的檢測數據,以便在未來重新挖掘以往的數據。采用高分辨質譜風險篩查還可以在沒有標準品的環境下進行初步篩查,降低實驗室采購標準品的成本。例如,Gao 等[28]采用了25 種不同性質的物質(含有機磷類農藥、氨基甲酸酯類農藥、四環素類抗生素、磺胺類抗生素、喹諾酮類抗生素、酰胺類除草劑、三嗪類除草劑和殺菌劑等),驗證了并聯固相萃取方案,解決了非靶向有機危害物流入、假陰性和假陽性的風險問題。所選用的質控物質如表1 所示。
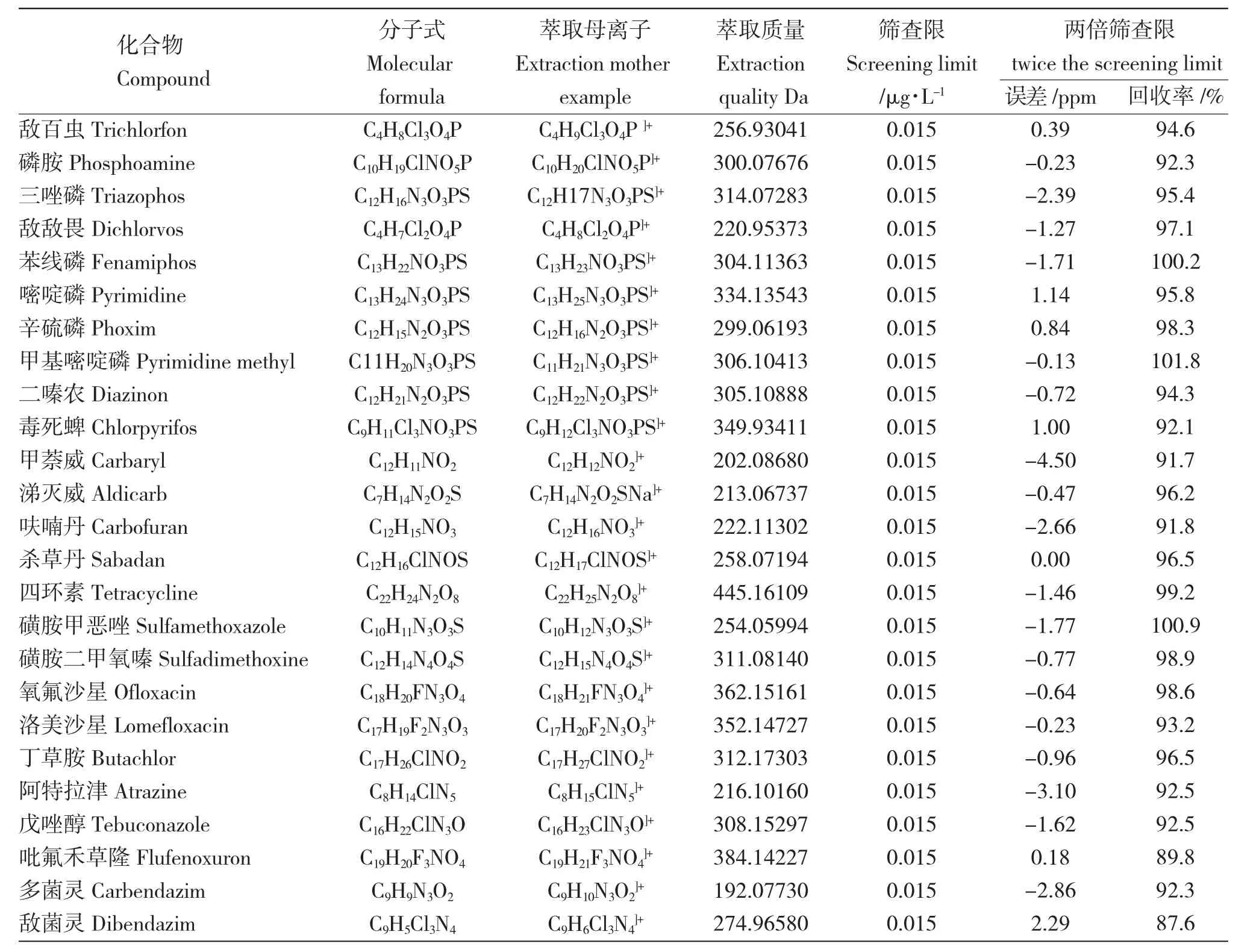
表1 并聯固相萃取方法中質控物質的篩查限及回收率Tab.1 Screening detection limit and recovery of quality control compounds with different properties in the parallel solid phase extraction
2.2.2 風險預警
利用高分辨質譜數據的追溯性,可保存平時的“陰性”水和底泥等環境樣本,在水產品發生風險時,可對高分辨質譜數據進行追溯。將已有風險的污染樣本A(圖2 左)對照陰性樣本B(圖2 右)。Gao等[28]建立了無差別無歧視的盲篩對比表格,分析得到信號值差異最大的幾組數據。由圖2 可知,樣本A 在5.80 min 時,一級質譜圖中有216.1009 Da 的響應,其具有二級譜圖(104.0023 Da、146.0218 Da、174.0539 Da、216.1048 Da)。而反觀樣本B,在同樣的出峰時間(5.80 min)卻沒有216.1009 Da 響應。進一步結合精確分子量、同位素豐度比等進行判斷,初步判斷出候選物質C8H14N5Cl(誤差為0.2 ppm)、C13H13NO2(誤差為-3.7 ppm)、C9H9N7(誤差為8.7 ppm)、C8H13N3O4(誤差為14.9 ppm)和C10H17N2O2S(誤差為-19.3 ppm)。根據二級質譜圖(104.0023 Da,146.0218 Da,174.0539 Da,216.1048 Da)裂解規律判斷碎片離子,判斷碎片離子為C8H15N5Cl+(216.1048 Da,誤差為3.8 mDa)、C5H9N5Cl+(174.0539 Da,誤差為-0.2 mDa)、C5H9N3Cl+(146.0218 Da,誤差為-26.2 mDa)和C2H3N3Cl+(104.0023 Da,誤差為1.3 mDa)。判斷污染物可能為阿特拉津。后續可根據風險預警的判斷結果再進行下一步的確證。待驗證正確后,將其加入信息數據庫共享,可為日后風險預警提供技術支持。
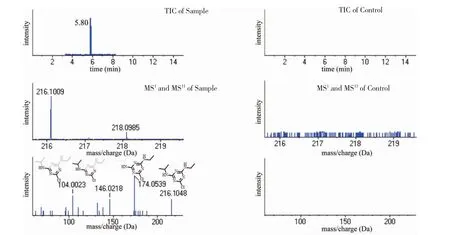
圖2 風險預警篩查得到的特異物質總離子流圖、一級質譜圖(MS1)及二級質譜圖(MSn)Fig.2 The total ion chromatogram,MS1 and MSn spectra of the compound obtained from risk warning screening
2.3 多方面確證
針對篩查出的有毒有害可疑風險物質,采取多譜圖結合的方式,如質譜+色譜+光譜+核磁等多譜圖驗證,解決假陽性問題。以并聯固相萃取高通量前處理為技術為基礎,結合LC-Q-TOF-MS 篩查技術,對東北地區部分池塘進行了污染物的高通量篩查,結果在哈爾濱某漁業養殖基地水源中篩查到有丁草胺的殘留。隨后又分析了此水環境中的鯉(Cyprinus carpio)樣品的數據,提取特征母離子312.17303 Da 的萃取離子流圖(XIC)(圖3-A),發現在7.77 min 時,有一個強度僅為40 的疑似丁草胺峰(圖3-B)。由于其MS1 豐度較低,信息依賴采集模式并沒有采集到其子離子(圖3-C)。
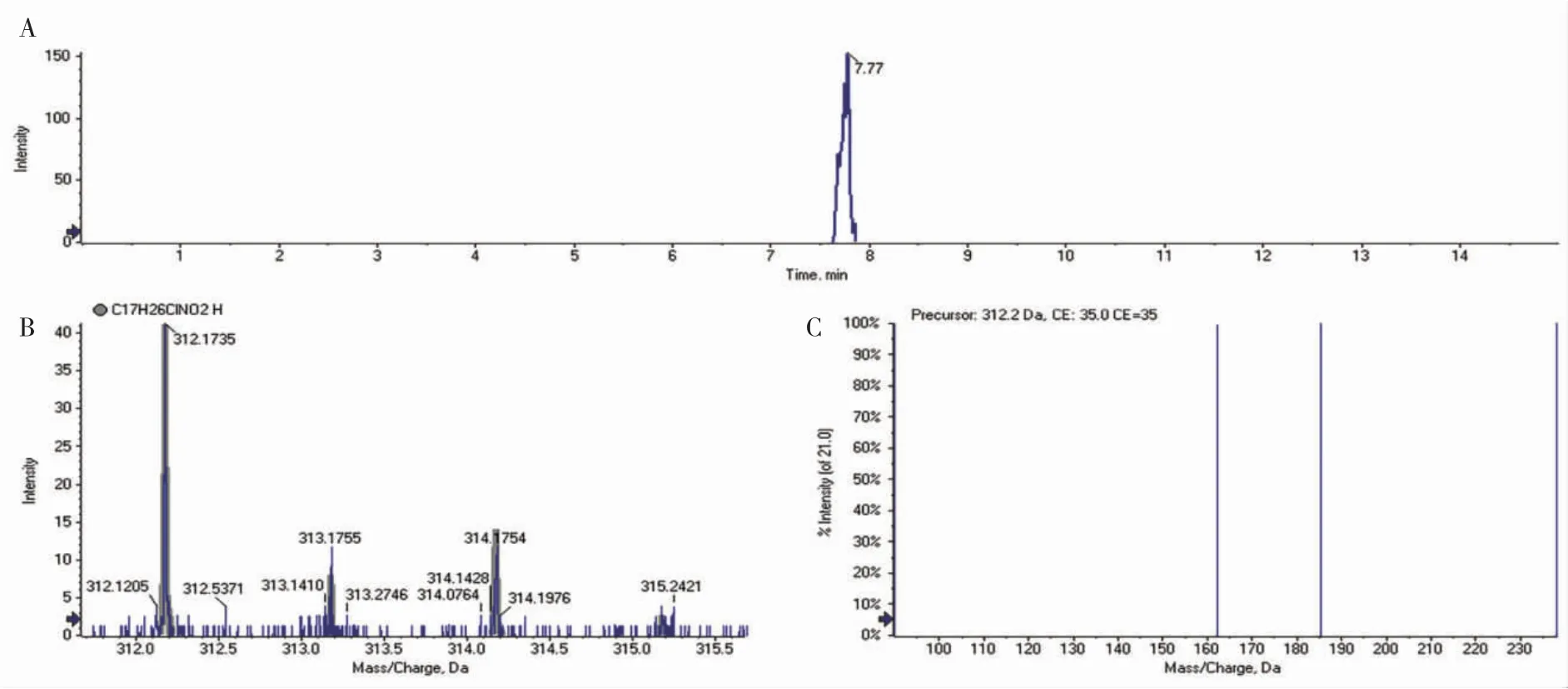
圖3 鯉中污染物的萃取離子流圖(A),MS1 圖(B)及MS2 圖(C)Fig.3 Extracted ion chromatogram(A),MS1(B)and MS2(C)of pollutants in common carp
為進一步鑒定該污染物,采用液相濃縮方式將7.50~8.00 min 的餾分進行多次收集濃縮(色譜的TIC 圖如圖4-A 所示),并進一步采用泵直接進樣的累加模式采集餾分的MS1(圖4-B)和MS2(圖4-C)譜圖。發現312.1735 Da、313.1771 Da 和314.1705 Da 等幾個MS1 特征峰,及238.0972 Da、162.1270 Da 和57.0713 Da 等幾個MS2 特征峰。對MS2 進行分析,通過理論分子模擬、碎片裂解規律,解析出了C13H17NOCl+(238.0993 Da)、C11H16N+(162.1277 Da)和C4H9+(57.0699 Da)三個MS2 碎片離子信息。根據《東北地區主要涉漁農藥及鑒別》圖書中第三章“涉漁農藥色譜-質譜圖集”報道[29],其指紋圖譜庫如圖4-E 所示。通過以上信息及自建的標準指紋圖譜庫比對,及根據歐盟對有機物的判定規則[30],其高分辨質譜的母離子加2.0 分,每個子離子加2.5 分。此化合物打分為2.0+2.5×3=9.5 分,已經超過其規定的分數。故可判定7.77 min 的312.1735 Da 物質就是丁草胺污染物(結構式如圖4-D 所示,解析結果如圖4 箭頭所示),證實該環境下魚肉樣品為陽性。
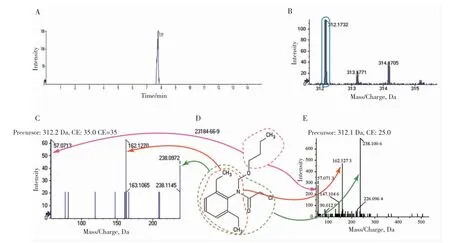
圖4 濃縮后污染物的色譜圖(A),MS1 圖(B),MS2圖(C),丁草胺結構式(D),圖書中丁草胺指紋圖譜庫(E)Fig.4 Chromatograms(A),MS1(B),MS2(C)of compound after concentration,structure of butachlor(D),and library of butachlor(E)
其中篩查及確證技術在其他農產品領域也有部分應用。謝瑜杰等[31]采用液相色譜-四極桿飛行時間質譜快速篩查蔬菜中農藥,孟志娟等[32]結合氣相色譜-四極桿-飛行時間質譜篩查水果蔬菜中農藥;王思威[33]和雷汝清[34]等分別應用高分辨質譜非靶向篩查了蜂巢蜂蜜中的農藥及西紅柿中殺菌劑等有機危害物。采用實驗室進行風險篩查及預警進而進行多方面驗證可互補現場快檢方案。
2.4 數據追溯及風險控制
可追溯的數據可盡可能地控制風險。檢測過程中保留經典高分辨質譜數據(含信息依賴性掃描及全掃描圖譜)十分必要,可以保留“陰性”樣本,達到數據可追溯的目的。由于水產品富集倍數不足的原因,可能當時無法篩查出風險物質丁草胺。當發現其中的一個樣本中含有某個風險物質時,可以利用高分辨質譜數據的可追溯性針對性地觀察某個物質。如2.3 節中已知水環境中含有丁草胺的殘留,于是追溯其環境和水產品中丁草胺的含量,發現其環境中鯉7.77 min 時的312.1735 Da 物質,并對其進行富集檢測及分析、驗證。目前受到檢測技術發展的制約,可能造成檢測的假陰性,原因可能是水樣品經過了固相萃取大體積的富集濃縮,很容易將污染物富集從而提高了濃度;水環境樣品的凈化提取更為簡單,而水產品的細胞破碎困難,使得常規方法不能有效提取污染物,造成提取效率低、回收率低;水樣品基質簡單,更容易從眾多的雜峰中找到污染物,而水產品肌肉基質復雜,含雜質多,采用非選擇性的前處理手段會影響除草劑的分離,會將其低豐度的特征峰(如312.1735 Da、313.1771 Da 和314.1705 Da)誤認為雜質。如今受到檢測技術發展的瓶頸,會導致樣品出現“假陰性”,因此隨著檢測技術的飛速發展,保留“原始數據”的文件十分必要,可為后期追溯及溯源提供保障。
2.5 提升檢測科研水平
目前水產品檢測技術中針對呋喃西林的代謝物氨基脲(SEM)的檢測往往存在一定的假陽性風險。隨著我國科研水平的提高,檢測技術也在不斷完善。王建[13]研究認為,采用2-硝基苯甲醛作為衍生化試劑很可能無法準確鑒定不同來源樣本中的SEM,采用硝基糠醛作為生物標示物,可有效避免檢測中非呋喃西林源和呋喃西林代謝產生的SEM 的餛淆,解決SEM 檢測過程中的假陽性問題,當標準得以驗證后亦可對標準進行更新,解決標準滯后性的問題。
隨著我國檢測科研能力的提高,會逐步解決檢測過程中出現的假陰性、假陽性及標準滯后性等問題。針對現在主動用藥的監管已經成熟的條件下,對“被動”用藥的研究還需要更加深入,要理清藥物的來源,查清漁用投入品、飼料、非藥品類漁用投入品、水源、底泥、周邊環境等。通過快檢來快速篩查以達到加快現場監管,通過高通量篩查對“被動”用藥實現風險篩查,利用多方面信息確證有機危害物,最終實現風險防控。
3 結語與展望
本文提出了水產品中有害物殘留檢測所遇到的挑戰,展望了未來水產品檢測的發展新思路。這種以現場快檢初步定性定量,并在實驗室進行高通量非靶向風險篩查及預警,結合多方面確證有機危害物,可以做到數據可追溯、風險可控制,終端可快檢,實驗室檢測準,具有時效性強、可覆蓋面廣的優點,能在一定程度上保障我國水產品質量安全。目前水產品質量安全還任重道遠,高端分析儀器國產化、高精尖人才本地化,這都需要涌現出一批年輕力量。相信隨著檢測科研的投入,未來檢測手段將有新的突破。在目前互聯網的大潮中,可推進“互聯網+水產品質量安全”,可實現實時更新我國水產品質量安全情況,建立信息公開化的水產品有機危害物數據庫,構建信息追溯平臺,以實現新時代中國綠色水產品的美好愿景。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
