
鋰金屬負極的挑戰與改善策略研究進展
2021-03-05 12:29:54劉凡凡張志文葉淑芬姚雨余彥
物理化學學報 2021年1期
關鍵詞:生長
劉凡凡,張志文,葉淑芬,姚雨,余彥,2,*
1中國科學技術大學,中國科學院能量轉換材料重點實驗室,材料科學與工程系,合肥微尺度物質科學國家研究中心,合肥230026
2中國科學院潔凈能源創新研究院,遼寧 大連 116023
1 引言
隨著電子產品的蓬勃發展和汽車電動化的進展,社會對電池儲能器件的比能量需求日益增高,近20年來,以石墨為負極,鈷酸鋰(LiCoO2)和磷酸鐵鋰(LiFePO4)以及三元材料為正極的鋰離子電池已經得到廣泛的應用和發展1-4。然而,由于傳統石墨負極材料較低的理論比容量(372 mAh·g?1)以及較高的電壓平臺,造成傳統鋰離子電池無法進一步突破其比能量瓶頸(260 Wh·kg?1)5。因此我們需要進一步去探索具有高理論比容量和低電極電勢的負極材料,從而在電池材料體系上使電池達到更高的比能量6,7。近些年來,由于鋰金屬具有高理論比容量(3860 mAh·g?1)和低電極電勢(?3.04 VvsSHE (standard hydrogen electrode))等特點,鋰金屬負極從而得到電池研究學者們的廣泛關注。將鋰金屬負極匹配過渡金屬氧化物正極(LMO)構成鋰金屬電池時,其比能量可以提升到約440 Wh·kg?1。在鋰硫(Li-S)和鋰氧(Li-O2)電池體系中,其比能量可以進一步分別達到~650和~950 Wh·kg?1,其比能量對比結果如圖1a所示8?10。
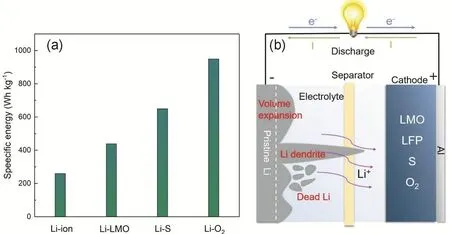
圖1 (a)鋰離子電池和鋰金屬電池的比能量對比;(b)鋰金屬電池中鋰負極存在的問題Fig. 1 (a) The specific energy comparison between Li ion battery and Li metal battery;(b) the main problems of Li anode in Li metal battery.
然而,鋰金屬作為電池負極的應用仍然面臨著諸多問題。在上個世紀七八十年代,鋰二次電池便是使用金屬鋰作為負極,但是由于其存在嚴重的安全問題而被棄用11,12。其中,鋰枝晶便是鋰金屬電池使用過程中的主要問題所在,在電池多次充放電后,鋰金屬表面會不斷生長出鋰枝晶,大量枝晶對隔膜具有較大的應力導致隔膜被刺穿,從而引發電池短路、著火、產氣爆炸等熱失控問題13,14。此外,鋰金屬的無限體積膨脹也會導致其鋰電極粉化、電解液消耗、大量產氣等各種問題,這些問題使鋰金屬負極難以實現商業化應用15。然而,電池研究學者們從未放棄這樣的高比能量負極材料,一直對這樣的“優秀但淘氣”的負極材料進行不斷的改性和優化16,17。本文主要對鋰金屬的問題,鋰枝晶成核生長原理以及鋰金屬改善策略進行了綜述,以促進對鋰金屬負極的進一步認識,推動高比能量鋰金屬電池的發展。
2 鋰金屬問題和枝晶生長模型
2.1 問題與挑戰
由于鋰金屬在堿金屬中具有最負電位和最低密度(0.53 g·cm?3),因而促進了其在高比能量鋰二次電池中的應用前景。然而,鋰金屬具有較高反應活性,容易與有機電解液反應生成Li2CO3、LiOH、Li2O、Li3N、LiF等無機產物18和ROCO2Li、ROLi、RCOO2Li (R是烷基官能團)等有機產物19。這些反應會導致鋰金屬和電解液的利用率降低,并會伴隨著大量的氣體產生,容易引發鋰金屬電池的安全隱患。此外,鋰金屬的高活性和其表面SEI的鋰離子擴散能壘較高會促進鋰枝晶的形成。如圖1b所示,鋰金屬的不均勻沉積和枝晶生長會引發以下主要問題20,21:1)鋰的無限體積膨脹;鋰金屬不同于石墨,硅等嵌入型或合金類負極,它是一種無基體轉化型負極,石墨和硅的體積膨脹分別是10%22和400%23,而鋰負極的體積膨脹是無限的,導致沉積鋰的形貌結構呈現多孔疏松的狀態。2)死鋰的產生;鋰的無限體積膨脹和枝晶均會造成鋰表面結構多孔疏松,經過多次充放電循環后,表面不穩定的鋰會逐漸粉化并脫落下來從而失去電活性,從而產生大量死鋰。3) SEI破裂和副反應增加;鋰枝晶的生長和死鋰的產生會導致鋰表面SEI破裂和重構,不斷的重構SEI需要消耗額外的電解液,造成副反應增加。4)極化電壓增大;鋰枝晶和死鋰導致鋰金屬表面多孔疏松,SEI的比表面積和厚度均會隨之增大,從而使Li+的擴散路徑增加,并且死鋰會導致表面阻抗增加,這些因素都會造成鋰金屬電池在多次循環后的極化電壓顯著增加。5)電池短路;鋰枝晶的不斷生長會造成其對隔膜的應力增加,最終會刺穿隔膜導致電池短路,從而引發電池熱失控等安全問題。綜上所看,鋰金屬的挑戰眾多,需要不斷的探索新的策略以解決其存在的諸多問題。
2.2 枝晶成核生長模型
鋰金屬負極在充放電循環過程中不斷進行著鋰的脫鍍反應,因此研究鋰枝晶的生長行為對發展穩定的鋰金屬負極具有指導性意義。下面對鋰枝晶生長模型進行總結以幫助理解鋰的成核和生長行為。
2.2.1 表面成核生長模型
在實驗研究中發現鋰金屬在充放電過程中很容易產生大量的枝晶,而鎂金屬作為負極的時候基本不會產生枝晶狀的形貌24,25。針對于這一實驗現象,Ling等26提出了由于鎂原子之間的Mg-Mg鍵結合相對于Li-Li能更大,因此在不同維度的自由能差更大,故而更傾向于形成三維或者二維的結構,很難形成不穩定的一維枝晶狀結構。Jaeckle等27進一步的研究表明了Mg在(0001)界面相對于Li和Na在(001)界面具有更快的表面自擴散系數從而形成了鎂金屬表面的無枝晶形貌。結合這兩方面研究工作可以得到表面能和擴散系數是影響鋰枝晶生長的兩個因素。當鋰表面擴散系數低時,鋰在SEI下成核并會持續聚集沿一維結構生長形成大量鋰枝晶,因此在鋰表面構建具有高表面能組分和高鋰擴散系數可以有效抑制鋰枝晶的生長。
2.2.2 電荷誘導生長模型
鋰離子的不均勻沉積容易導致該金屬鋰表面形成突起鋰核,Ding等28證實由于尖端效應鋰核尖端容易聚集大量的電子從而具有更高的局部電場,該局部更高的電場強度容易吸引更多的Li+聚集沉積生長成鋰枝晶。該研究者們基于能斯特方程進一步提出了添加陽離子對突起鋰核形成靜電屏蔽作用從而抑制鋰的尖端生長。在電解液中添加少量的金屬陽離子(M+)添加劑,其還原電勢為:
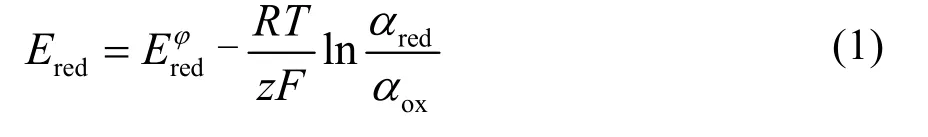
2.2.3 SEI擴散控制模型
在鋰金屬電池充電過程中,Li+從正極脫出,然后通過電解液中離子擴散,在負極鋰表面得到電子還原成為單質Li。Chazalviel等29建立雙極板模型,研究負極表面微區的濃度變化。根據菲克第二定律,并結合邊界束縛條件在負極表面建立如下的濃度梯度公式:
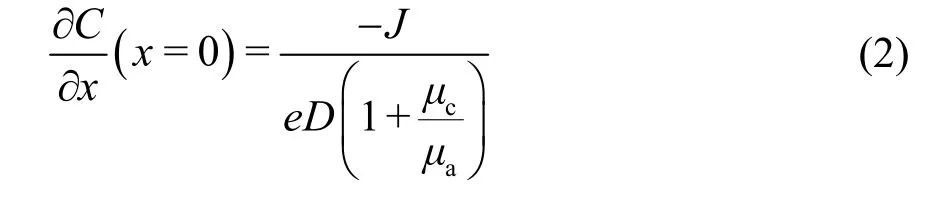
其中J是電流密度,e是電荷,D是雙極擴散因子,μc和μa分別是陰離子和陽離子的遷移數。當?C/?x <2C0/L,電解液中離子濃度達到穩態,其中C0是電解液中初始鋰離子濃度,L是極板間距離。當?C/?x >2C0/L,正負極表面離子濃度相差甚大,在負極表面微區鋰離子濃度很快降低為零并形成電負性空間電荷區,從而進一步吸引鋰離子生長為枝晶鋰,極化電壓顯著增大。這段鋰表面微區Li+消耗完的時間稱為過渡時間,又稱為“sand時間”,該時間值為:
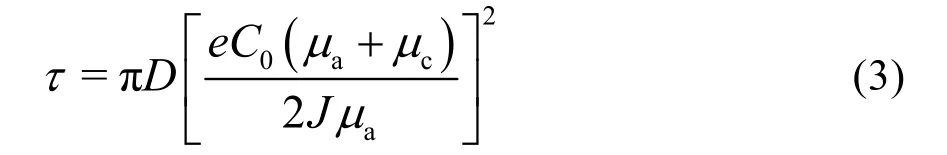
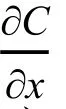
2.2.4 沉積-溶解模型
沉積-溶解模型主要是從熱動力學角度來解釋死鋰以及表面顆粒狀堆積鋰產生的過程和原由30。在鋰金屬電池充電過程中,鋰負極表面鋰沉積的主要過程如下:1) Li+遷移通過SEI薄膜沉積為鋰金屬,此時SEI薄膜未有損壞;2)在鋰沉積點的位置由于鋰對SEI薄膜機械應力的作用使此處有更高的離子電導率,則在此處鋰的沉積速度更快而致使鋰離子流不均勻,此外鋰表面的晶體缺陷和晶界也會引發鋰的連續不均勻沉積;3)由于應力的作用,沉積鋰的形貌會發生變形來緩解此處應力,此時鋰的沉積受限于沉積點曲面位置的鋰表面張力,SEI薄膜缺陷,鋰表面晶體缺陷和晶界;4) SEI薄膜層在鋰生長應力的作用下破損,鋰進一步生長為晶須狀鋰;5)彎折的晶須鋰在結點處會發生斷裂和溶解,致使產生顆粒狀鋰和死鋰,在高電流密度下或者低溫條件下更容易產生大量死鋰,這是源于這種條件下的晶須鋰更容易斷裂。當考慮到鋰枝晶的變形應力差是由表面張力引起的,其滿足以下公式:

其中ΔP是鋰枝晶變形應力差,γ是鋰的表面張力,R1和R2是對應曲面的曲率半徑。Yamaki等30通過計算模擬得到當ΔP/γ小于0.2 μm?1,可以抑制枝晶生長對SEI保護層的破壞。
除以上介紹的主要枝晶生長模型之外,還有相場模型31,32,異相成核模型33和空間電荷模型34等,基于對枝晶生長模型的深入了解會幫助研究者們進一步探索出解決鋰枝晶生長問題的策略。
3 三維導電骨架改善鋰金屬負極
鋰金屬負極由于其無基體轉化反應的特性而存在無限體積膨脹的問題,針對于此類問題,研究者們設計多種三維導電骨架用于預儲鋰基體,三維導電骨架不僅可以限制鋰的無限體積膨脹而且能夠均勻化鋰離子流,降低表面有效電流密度,從而抑制鋰枝晶的生長。以下對近些年主要研究的三維基體骨架策略進行總結分析以明白其設計原理和作用機理。
3.1 石墨烯復合基體
石墨烯在近年來的研究中已被廣泛應用于儲能電極材料體系中35-37,其主要原因在于石墨烯的高電子電導率,結構穩定,密度低和易摻雜等優勢38,39。基于這些優點,石墨烯也可以被應用于金屬鋰的結構基體中來優化鋰負極40-43。Lin等44使用具有親鋰性的分層還原氧化石墨烯(rGO)作為儲鋰基體,如圖2a,利用熱熔融的方法將液態鋰滲入到分層石墨烯基體中得到復合金屬鋰電極LirGO,該復合電極仍然具有3390 mAh·g?1的比容量。該復合電極中的石墨烯有以下作用:1)石墨烯可以作為一個穩定的金屬鋰限域骨架;2)熱處理后的石墨烯表面具有親鋰性的含氧官能團,能夠促進金屬鋰的均勻沉積;3)石墨烯層間表面可以作為一個穩定的人工SEI表面。在上述的設計優點下該Li-rGO復合電極表現出優異的電化學性能,在電流密度為3 mA·cm?2,脫鍍容量為1 mAh·cm?2的條件下Li-rGO展現出過電勢僅有80 mV的穩定循環,在和鈷酸鋰組裝的全電池中同樣具有更優的電化學性能。
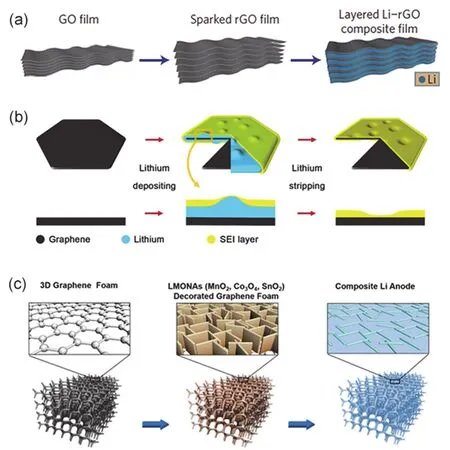
圖2 (a)分層還原氧化石墨烯復合鋰電極合成示意圖44;(b)氮摻雜石墨烯誘導鋰沉積過程示意圖45;(c)金屬氧化物納米片修飾石墨烯泡沫熔融復合鋰金屬46Fig. 2 (a) Schematic of fabricating layered rGO and Li composite electrode 44. (b) Schematic of inducing Li deposition via N-doped graphene 45. (c) Schematic of fabricating Li and graphene foam modified by metallic oxide nanoarrays 46.
Zhang等45利用MgAL-LDH作為模板,如圖2b所示,通過化學氣相沉積法制備出非堆疊的石墨烯薄片并將其涂覆在銅箔上應用于鋰的電沉積基體。由于其石墨烯片層具有較高的比表面積,其表面局部電流密度則很大程度上減小從而形成穩定的鋰沉積形貌和結構。并且該工作指出隨著鋰沉積在導電石墨烯片層上時形成石墨烯-沉積鋰-SEI的三明治核殼結構。當鋰脫出時其SEI仍然保持完整,未出現破損,體現其SEI的柔韌性和穩定性。該設計的電極結構具有調控鋰離子的均勻沉積和抑制枝晶鋰生長的作用,從而在電流密度為2 mA·cm?2,脫鍍容量為5 mAh·cm?2的條件下實現了93%的庫侖效率,并且在對稱電池中實現了800圈的循環壽命。
由于金屬氧化物能夠和金屬鋰進行氧化還原反應,故而具有優異的親鋰性,利用此特性Yu等46在石墨烯泡沫上分別修飾多種金屬氧化物納米片(如MnO2、Co3O4、SnO2)以促進熔融鋰進入到石墨烯泡沫骨架中得到復合金屬鋰電極,其過程如圖2c所示。其中,石墨烯泡沫骨架具有限制鋰的體積膨脹和穩定電解液/電極界面的作用,金屬氧化物納米片可以提高其骨架親鋰性,降低局部電流密度并且同時誘導鋰離子的成核和后續沉積過程。結合石墨烯泡沫骨架和金屬氧化物的優點,在電流密度為1 mA·cm?2,脫鍍容量為1 mAh·cm?2的條件下基于該復合金屬鋰電極(Grphene/MnO2基體)的對稱電池中可以穩定循環1600 h。
親鋰性的多孔石墨烯材料主要用于熔融鋰儲存的基體和鋰電沉積的集流體。除上所述方案外,關于石墨烯的基體應用于鋰金屬的研究仍然很廣泛,如雜原子摻雜的石墨烯對鋰沉積的影響47-49,石墨烯納米籠負載Au顆粒對鋰沉積的作用50,以及納米多孔石墨烯51應用于熔融鋰的基體等多種策略優化金屬鋰負極。
3.2 碳纖維復合基體
自支撐碳纖維基材料由于其穩定的三維骨架網絡,優異的電子導通性質和高比表面積等優點一直以來廣泛被應用為電極儲能材料52-54。由于碳纖維的三維結構可以降低有效電流密度,減小鋰成核過電勢,限制金屬鋰的體積膨脹,因而碳纖維基體也被廣泛應用于鋰金屬沉積的集流體,如石墨化碳纖維55,三維多孔碳纖維56,碳纖維布等57,58。
然而單純碳纖維的親鋰性比較差,因此需要對碳纖維進行表面修飾以促進熔融金屬鋰的滲入和調控鋰離子的均勻沉積。Yang等59通過焦耳熱方法在自支撐碳纖維上沉積了尺寸為40 nm的超細金屬Ag顆粒用于誘導鋰沉積行為,如圖3a所示,并率先提出了Ag顆粒可以提高碳纖維的親鋰性。實驗結果表明鋰可以在修飾Ag顆粒后的碳纖維骨架更加均勻地沉積,并且其成核過電勢減小到基本可以忽略不計,證實了Ag顆粒的修飾可以降低局部鋰離子流密度,減小局部電流密度,并作為鋰沉積的成核和生長位點,實現了對金屬鋰沉積的均勻調控并抑制了鋰枝晶的生長。受到此設計策略啟發,Zhang等60也進一步在碳纖維表面電沉積珊瑚狀Ag顆粒層,再通過熔融鋰的方式制備了復合金屬鋰電極,并通過第一性原理計算證實了Ag(?2.07 eV)相對于碳纖維(?1.14 eV)對鋰具有更高的結合能值,從而實現了對鋰離子的均勻調控。此外,具有相似性質的Au納米顆粒61也被用于修飾碳纖維來調控鋰離子流,同樣取得了無枝晶鋰生長的結果。
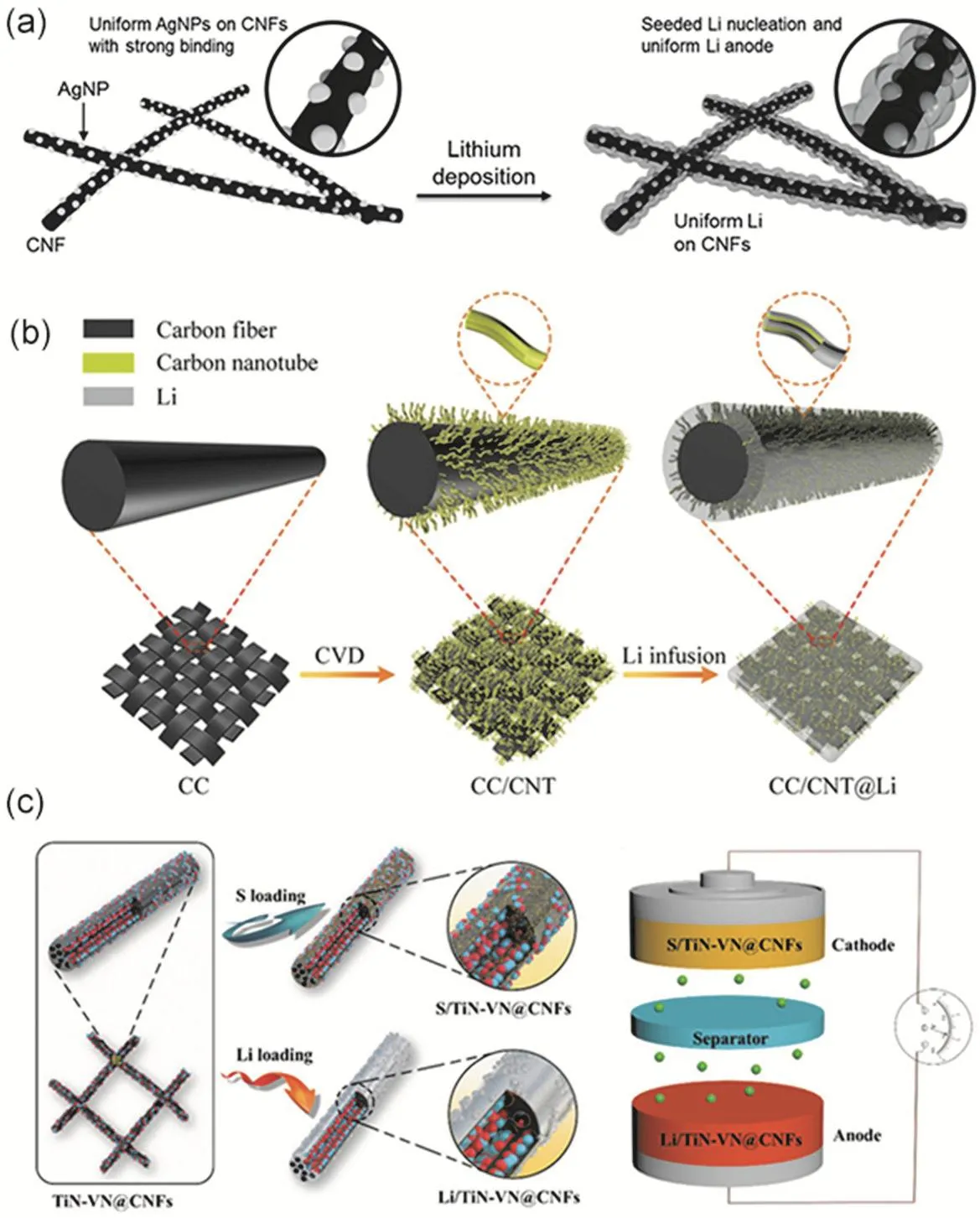
圖3 (a) Ag顆粒修飾碳纖維對誘導鋰沉積行為示意圖59;(b) Li@CC-CNTs復合電極合成過程示意圖62;(c) TiN-VN@CNFs應用于硫正極和鋰負極過程和作用示意圖63Fig. 3 (a) Schematic of inducing Li deposition via Ag nanoparticle modifying carbon nanofibers (CNFs) 59. (b) Schematic of fabricating Li@CC-CNTs composite electrode 62. (c) Schematic of TiN-VN@CNFs applied for Li anode and sulfur cathode 63.
碳纖維布由于其在高溫滲鋰過程中仍然能夠保持穩定的纖維結構而在近期廣泛應用于復合鋰金屬負極的載體58,64。Liu等62通過化學氣相沉積在碳纖維表面修飾一層碳納米管,并通過高溫滲鋰實驗證明這些碳納米管不僅能夠促進熔融鋰滲入到纖維骨架中制備得到復合金屬鋰電極,而且能夠調控鋰離子在其表面均勻成核生長,如圖3b所示。該復合金屬鋰電極在鋰脫鍍的實驗過程中展現了無枝晶生長的均勻厚度變化,并電流密度為5 mA·cm?2,脫鍍容量為1 mAh·cm?2的條件下實現了500 h的穩定循環。此外,SnO265、RuO266、Co3O467、ZnO68,69等金屬氧化物也被報道提高碳纖維的親鋰性并調控鋰離子的沉積過程。為了進一步推進復合鋰金屬負極的實際應用,Go等70在碳纖維布上構建了納米裂紋以提高基體親鋰性,然后通過高溫熔融的方式大規模制備了碳纖維和鋰金屬的復合電極并實現了穩定的電化學脫鍍性能。
鋰硫電池中正極硫的多硫化物溶解穿梭效應和負極金屬鋰的枝晶生長問題是限制鋰硫電池應用的主要問題71,72,針對于這一問題,Yao等63設計一種負載TiN-VN異質結的柔性碳纖維(TiNVN@CNFs),將其分別與硫和金屬鋰復合,并用于Li-S電池的正極和負極材料(如圖3c所示),同時解決了正極多硫化鋰的溶解穿梭問題以及負極金屬鋰的不均勻沉積問題,實現了高比能量與長循環壽命兼備的鋰硫全電池。近期研究工作中設計合適的多孔極性電極材料同時用于解決鋰硫電池中的正極硫和負極鋰的問題是一種受到關注的研究策略73-76。
3.3 多孔金屬復合基體
由于高電子電導率和結構穩定性,金屬基體常被應用于負載電極材料的集流體77-80,故而三維多孔金屬基體也被探索應用于預儲存鋰的基體或者鋰電沉積的集流體以限制鋰金屬的無限體積膨脹和調控鋰離子的沉積行為81-85。Chi等86率先提出了將泡沫鎳應用于預儲存鋰的三維基體,由于泡沫鎳的高溫穩定結構和高電子電導率等特點,如圖4a所示,該研究者在加熱溫度為400 °C的條件下將熔融金屬鋰滲入到泡沫鎳的三維多孔骨架中構成泡沫鎳和金屬鋰的復合電極(Li-Ni composite)。該復合金屬電極在先脫出5、10、15 mAh·cm?2和重新沉積15、20 mAh·cm?2表現出均勻的厚度變化(該復合電極中1 mAh·cm?2的鋰對應的厚度為50 μm)以表現出該復合金屬電極的穩定性,其泡沫鎳表面的突起結構能夠起到調控鋰離子沉積作用,抑制鋰枝晶的生長。此外,該Li-Ni復合電極表現出優異的電化學性能,在對稱電池中電流密度為1、3、5 mA·cm?2的條件下表現出超過100圈循環的電化學穩定性,并且該復合電極在不同電流密度下的Li4Ti5O12和LiFePO4全電池中表現出更高的容量和優異的穩定性。經此設計策略啟發,泡沫銅82,87-90和泡沫鎳91-94及基體修飾策略進一步被開發和探索。
納米線結構材料由于其較高的比表面積和一維結構能夠構起到限域鋰和均勻化離子流等作用。Yang等95通過氧化還原法在Cu集流體表面構筑三維亞微孔銅納米線應用于鋰沉積儲存的集流體,其制備過程如圖4b所示,研究表明集流體表面的電化學活性區域面積決定著鋰儲存在三維集流體中的百分比值?:對于普通Cu集流體其?值為0,對于泡沫銅其?值為81%,意味著有19%的鋰沉積在三維骨架外面,對于三維銅納米線集流體其?值為98%,表明鋰基本全部沉積在集流體骨架中。該集流體在0.2 mA·cm?2的電流密度下表現出600 h的穩定電化學脫鍍循環曲線,并且在完成容量為2 mAh·cm?2的鋰的沉積/脫出過程后仍然能夠保持穩定的納米線結構。受此研究工作啟發,在集流體表面一個設計高電化學活性面積,多孔結構的思路廣泛用于調控鋰離子流的沉積和儲存鋰金屬,如三維垂直Cu微通道結構96,親鋰性CuO-Ni納米線結構97,三維多孔銅等98。
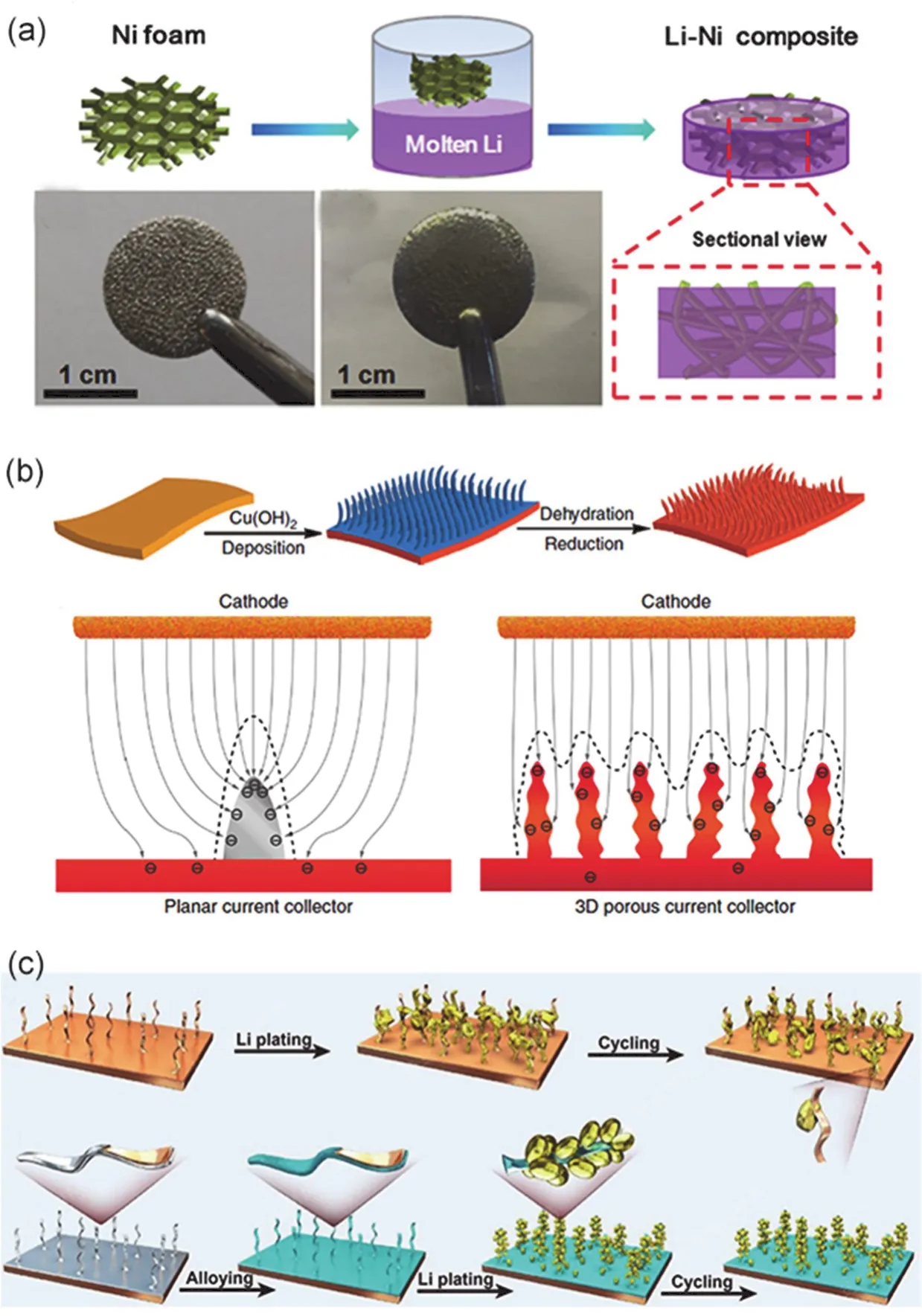
圖4 (a) Li-Ni復合鋰負極的合成示意圖和電極光學照片86;(b)多孔Cu納米線的合成示意圖,Cu集流體和多孔銅納米線集流體對鋰離子流調控作用示意圖95;(c) Cu纖維和包覆Al層Cu纖維對鋰誘導成核作用示意圖99Fig. 4 (a) Schematic of synthesizing Li-Ni composite and the corresponding optical photograph 86. (b) Schematic of fabricating Cu nanowires and the effects of regulating Li ions flux between planar and 3D porous Cu 95. (c) Schematic of inducing Li nucleation between Cu nanofibers and Al coated Cu nanofibers 99.
與鋰形成合金化合物的材料一般能夠降低鋰成核過電勢和誘導鋰成核。為了更好的控制鋰的成核過程,Ye等99首先在銅集流體表面構筑了銅纖維結構用于鋰沉積的骨架(3D Cu),然后通過磁控濺射的方法在銅纖維表面沉積了一層Al層(D Cu@Al),該Al層在鋰電沉積時可以和鋰形成一層LiAl合金層用作于鋰金屬的成核和生長,其過程如圖4c所示。研究表明鋰在3D Cu@Al表面沉積的形貌呈結節狀而非針狀,體現出LiAl合金層對鋰沉積均勻調控的作用。研究展示3D Cu@Al在對稱電池中(J= 0.5 mA·cm?2,C= 1 mAh·cm?2)展現出1700 h的穩定循環性能,在LiFePO4全電池中展現出250圈的穩定循環,而純Cu和3D Cu集流體的容量則快速衰減,表明LiAl合金層不僅具有補鋰作用而且能夠通過引導鋰的成核而抑制鋰枝晶的生長,促進鋰離子的均勻沉積。除LiAl合金外,LixSi100,LixZn101,LixSn102等與鋰形成合金化合物均能夠起到誘導鋰成核和促進鋰離子快速傳輸的作用。
綜上可以得知對于金屬集流體的主要設計策略集中于兩方面,一是設計多孔結構提高表面電化學活性面積提高儲鋰的空間,二是合金層或親鋰性化合物修飾金屬基體,起到誘導鋰成核生長和均勻化鋰離子流的作用。
3.4 粉體鋰金屬負極
以上幾種復合金屬基體主要集中于三維自支撐骨架,在本部分主要討論粉體材料用于儲存和保護鋰金屬負極。粉體材料相對于三維自支撐骨架的優點主要在于其大面積合成制備工藝更加簡單從而可以規模化制備復合鋰金屬負極。如圖5a所示,Wang等103首先通過噴霧干燥的方法合成碳納米管(CNTs)團簇球,然后通過高溫滲鋰的方式將適量熔融鋰滲入到CNTs粉體基體構成Li-CNTs復合粉體負極,最后將該粉體負極壓在銅集流體表面以用于電化學性能測試。由于CNTs的親鋰性,鋰完全浸入到碳納米管團簇球基體中,其中鋰的質量含量為60%。該復合電極Li-CNTs應用于對稱電池中,在電流密度為0.5 mA·cm?2,鋰脫鍍容量為20 mAh·cm?2的條件下能夠穩定循環400 h,而純鋰負極在循環5 h后則發生了短路現象。同樣地,Li-CNTs與LFP (LiFePO4)正極組成全電池后,在1C(160 mA·g?1)的電流密度下能夠穩定循環300圈,而純鋰負極在120圈后則發生快速的容量衰減。研究表明Li-CNTs的比表面積仍然有55.6 m2·g?1,是純鋰負極的三個數量級,其較大的比表面積使其電極表面局部電流密度降低,并且CNTs團簇球能有效地抑制鋰的體積膨脹從而抑制了鋰枝晶的生長和死鋰的產生,帶來了更優異的電化學性能。
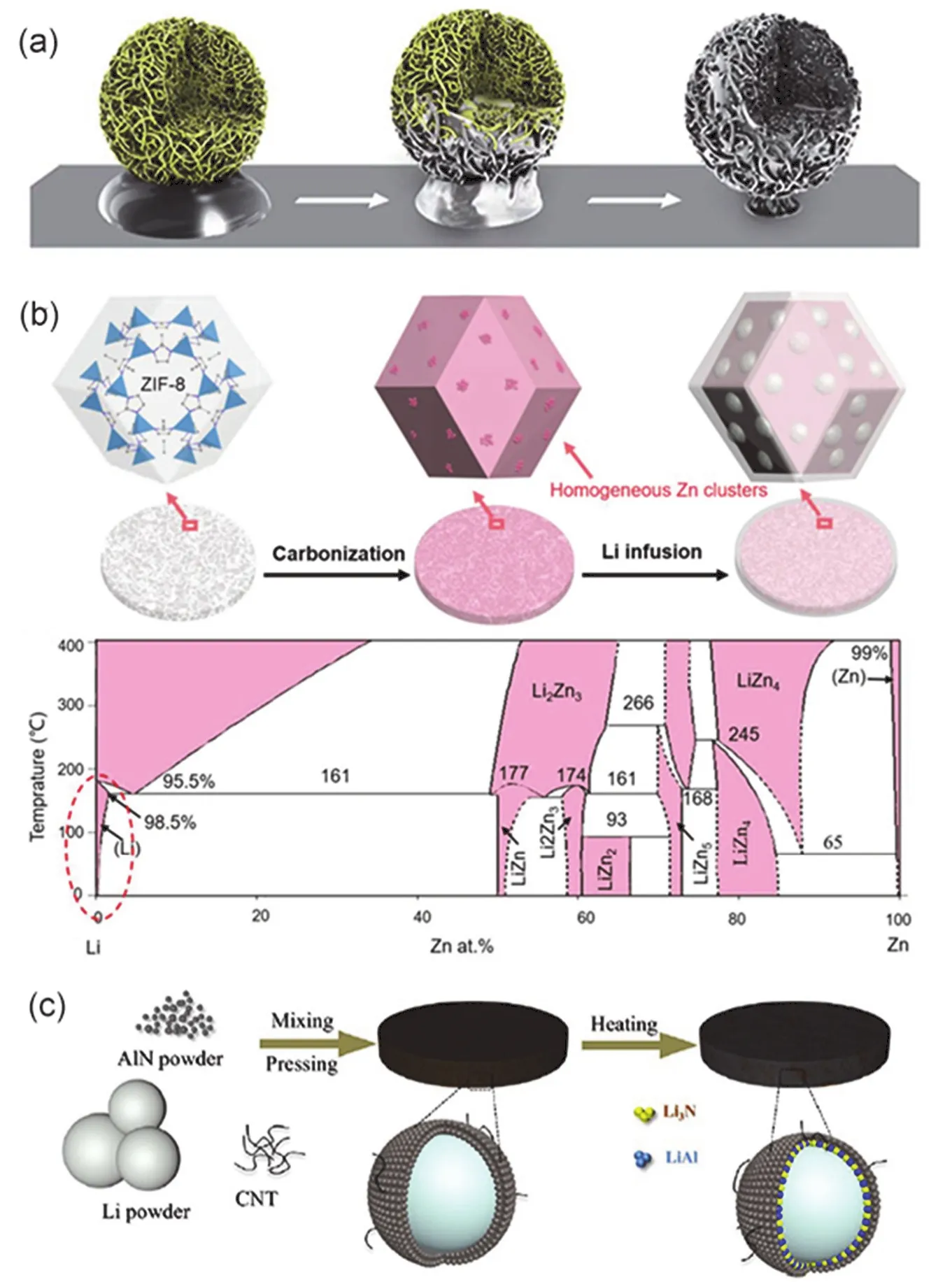
圖5 (a)碳納米管團簇球熔鋰過程示意圖103;(b) ZIF-8基碳材料熔鋰示意圖和Li-Zn二元相圖106;(c) LAN電極的制備過程示意圖109Fig. 5 (a) Schematic of infusing molten Li into CNTs cluster 103. (b) Schematic of Li and ZIF-8 based carbon material composite electrode and the corresponding Li-Zn binary phase diagram 106. (c) Schematic of fabricating LAN electrode derived Li powder, AlN and CNT powder 109.
金屬有機框架材料(MOFs)由于其獨特的配位方式和形貌結構在近些年廣泛被用于催化和儲能材料,其中ZIF-8是一種陽離子為Zn2+、有機配位鏈為二甲基咪唑的典型MOFs材料104,105。Zhu等106率先使用了碳化的ZIF-8材料(cMOFs)作為儲存鋰的基體,如圖5b所示,通過高溫滲鋰的方式將熔融鋰滲入到cMOFs的基體中制備成復合粉體鋰負極Li-cMOFs。cMOFs中具有親鋰性的Zn元素可以與鋰形成合金從而誘導熔融鋰滲入到cMOFs基體中,并且通過Li-Zn二元相圖可以知道在Zn含量為0.5% (w)的時候能形成Li-Zn固溶層,可以作為一個緩沖層而能夠有效地降低鋰沉積過程中的成核過電勢。得益于cMOFs的多孔限域效應和LiZn合金誘導作用,粉體復合電極Li-cMOFs對稱電池在電流密度為1 mA·cm?2,鋰脫鍍容量為1 mAh·cm?2的條件下能夠穩定循環700 h。此外,其他MOF類材料如ZIF-67107,MOF-199108也被用于均勻調控鋰的沉積行為。為了進一步得到離子和電子電導均優的復合鋰負極,Zhang等109將適量AlN粉末和球狀Li粉混合在一起,再加入少量的碳納米管壓成電極片,在150 °C條件下加熱2 h形成LAN復合鋰負極,其合成示意圖和作用機理如圖5c所示。其中和Li接觸的AlN反應生成高離子電導率Li3N和親鋰性的LiAl合金層,分別起到提高離子擴散動力學和誘導鋰成核沉積的作用,其外層高電子電導率的AlN和CNT提高了復合鋰負極的電子電導,減小了其極化電壓。此外,此種核殼粉體鋰負極結構不僅能夠限制鋰的體積膨脹而且能夠均勻分散鋰離子流密度從而抑制枝晶的生長,得益于這些優勢,該復合電極在鋰對稱電池、LFP全電池和S@PAN(硫@聚丙烯腈)全電池中均表現了穩定的電化學性能。
通過以上幾種典型的粉體鋰金屬負極材料設計策略我們可以知道粉體的多孔性和親鋰性質是其材料結構中的重要因素,并且可以通過設計合理的材料成分和結構來構建高離子擴散/電子傳輸動力學鋰負極。三維基體在解決了鋰無限膨脹等問題的同時也仍然面臨著一些問題,如其多孔和納米結構增加了副反應,骨架基體一定程度上降低了鋰的比容量等。為了了解不同基體對鋰脫鍍電化學過程的影響,四種主要基體構成的鋰負極對稱電池性能總結如表1所示。
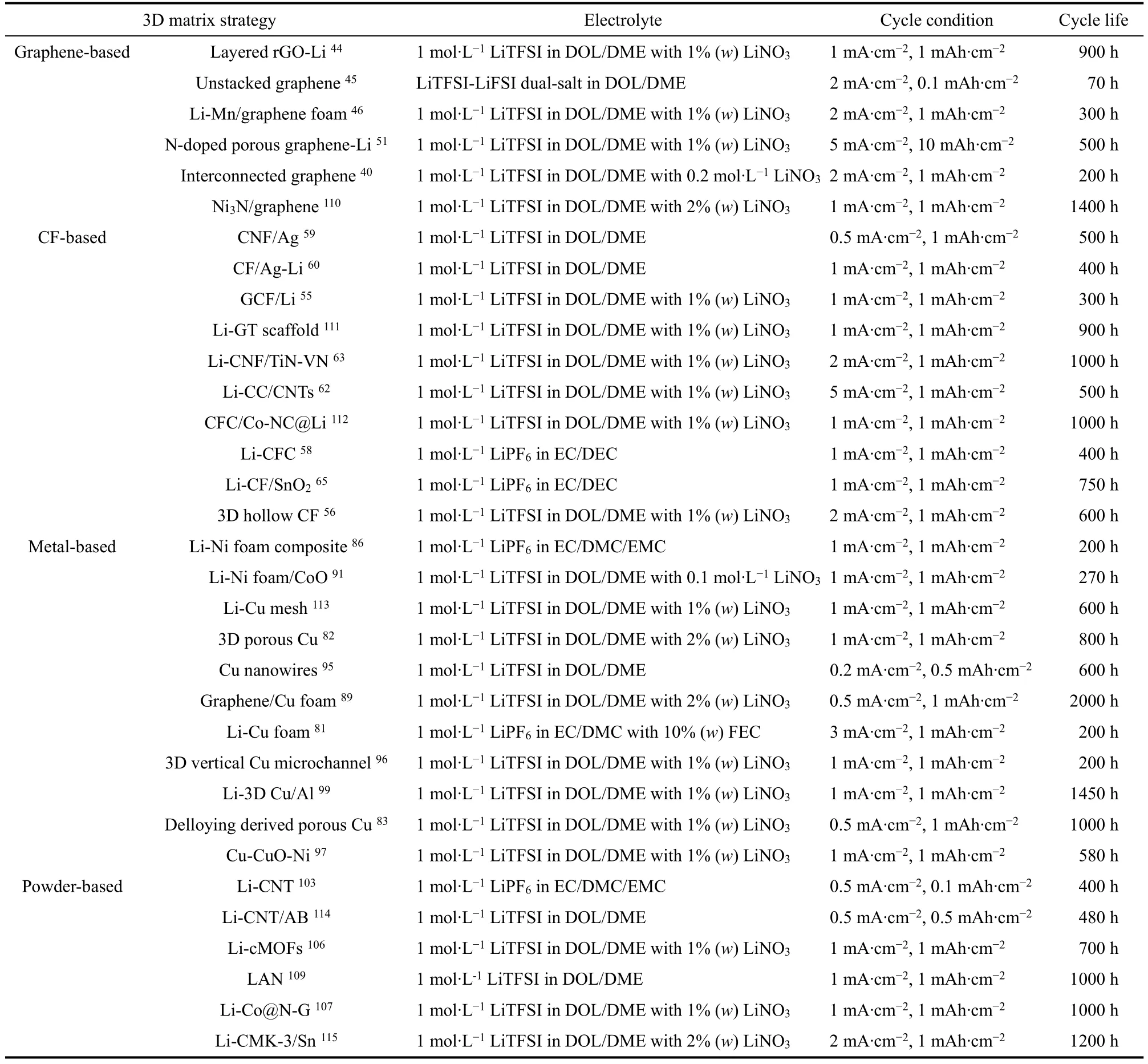
表1 不同基體鋰負極的對稱電池性能對比Table 1 The cycling performance comparison of Li symmetrical cells with different matrix.
4 電解液及添加劑改善鋰金屬負極
有機電解液是鋰離子/鋰金屬電池中的“血液”,負責正負極中鋰離子的傳輸,有機電解液由于其有限的電化學穩定窗口,一般會與電極材料發生化學反應生成固/液界面,即SEI薄膜。有機電解液和其中的添加劑一般會參與形成溶劑化鋰離子的殼層,在鋰金屬電池中,溶劑化殼層會參與反應形成鋰表面的SEI。因此,通過調控電解液成分、濃度和添加劑能夠有效地改變鋰金屬表面SEI的成分和調節鋰離子的沉積行為從而抑制鋰枝晶的生長116-124,并且對電解液的改善策略能夠進一步規模化應用到高電量鋰金屬電池中。為了進一步認識電解液對鋰金屬電池的影響,以下總結了幾種常見保護鋰負極的電解液優化策略,主要包括提高鋰鹽濃度,穩定SEI和誘導鋰沉積的添加劑策略。
4.1 高濃鋰鹽電解液
目前商業化鋰離子電池體系中電解液鋰鹽是LiPF6,溶劑為碳酸酯類體系,鋰鹽濃度一般處于1 mol·L?1左右,此種電解液體系在鋰離子電池中比較穩定,并且在此濃度附近電解液具有最優的離子電導率和液體流動性。而在鋰金屬電池體系中,研究者們發現通過提高電解液中鋰鹽濃度能夠有效地抑制鋰負極枝晶的形成123,127-129。在有機電解液體系中,鋰鹽中的鋰離子和有機溶劑形成溶劑化鋰離子,其陰離子也參與溶劑化殼層的形成,當鋰離子在鋰金屬表面沉積時,溶劑化鋰離子首先在SEI層進行去溶劑化的過程然后在鋰金屬表面發生沉積,此時溶劑化殼層還原分解參與形成SEI的成分130-132。電解液中溶劑分子和陰陽離子作用關系如圖6a所示126,當提高電解液鋰鹽濃度時,鋰離子周圍具有更多的溶劑化分子,并且自由陰離子能夠參與到形成鋰離子的溶劑化殼層,當稀釋高濃電解液時,自由陰離子參與形成的溶劑化鋰離子能夠仍然保持,因此構建合適濃度的高濃電解液能夠通過調節其溶劑化殼層的成分而進一步調節鋰負極表面SEI的成分從而抑制鋰枝晶的生長。Qian等133率先使用4.0 mol·L?1LiFSI/DME高濃度電解液體系應用于鋰金屬電池,相比于低濃度電解液,高濃體系Cu箔表面沉積的鋰形貌呈現穩定致密的塊狀,而在低濃體系中Cu箔表面呈現細長的枝晶狀。并且高濃體系的庫侖效率在1 mA·cm?2的電流密度下循環1000圈后仍然有99.1%。當該高濃體系電解液應用在Li對稱電池中,在10 mA·cm?2的電流密度下能夠穩定循環6000圈。而LiFP6鋰鹽中的陰離子基本不參與形成穩定LiF,并且酯類溶劑體系會促進表面更多的Li2CO3產生,從而使鋰枝晶很容易產生,導致庫侖效率較低。
Fan等126使用了有機溶劑為EC/DMC (碳酸乙烯酯/碳酸二甲酯),鋰鹽濃度為10 mol·L?1LiFSI的電解液體系應用于全氟化高壓鋰金屬電池。如圖6b所示,其中高濃度FSI?陰離子不僅在鋰負極表面還原形成富F的SEI層而且在三元正極材料LiNi0.6Co0.2Mn0.2O2(NCM622)表面氧化形成了含F的正極/電解液界面(CEI)。具有全氟化界面的NCM622鋰金屬全電池可以穩定充電至4.6 V,遠高于普通電解液體系NCM622的4.2 V或者4.3 V的充電截止電壓,如圖6c所示,10 mol·L?1高濃電解液體系在充電截止電壓為4.6 V的條件下能夠穩定循環100圈并保持99.1%的庫倫效率,同時在Li-Cu電池體系中能夠保持99.3%的庫倫效率穩定循環250圈,表明該電解液體系形成的氟化界面能夠抑制枝晶鋰的形成并提高鋰負極利用率。此外,如圖6d所示,通過分子能級對比發現LiFSI相對于EC和DMC具有更低的LUMO (最低非占據分子軌道)能級從而更容易在Li表面還原分解,LiFSI相對于EC和DMC具有更低的HOMO (最高分子占據軌道)能級從而具有更好的高壓穩定性。
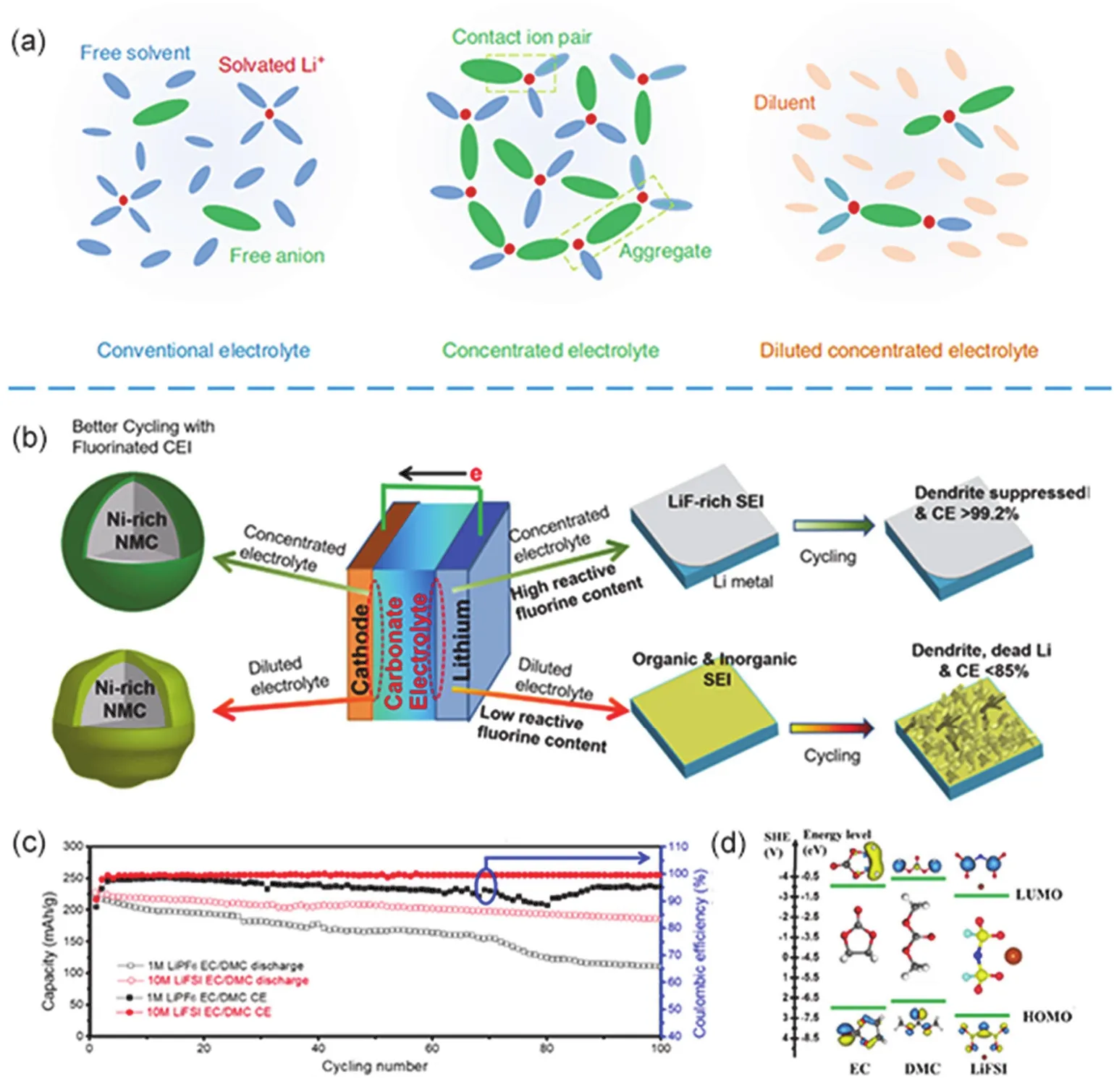
圖6 (a)普通電解液,高濃電解液,和稀釋后的高濃電解液中的溶劑分子和陰陽離子作用示意圖125;(b)全氟化高壓鋰金屬電池的正負極界面作用示意圖;(c)全電池循環性能對比圖,(d) EC、DMC和LIFSI的HOMO和LUMO能級對比圖126Fig. 6 (a) The effect schematic between solvent molecule and ions in conventional, high-concentrated and diluted concentrated electrolyte. (b) Schematic of the effect of highly fluorinated interphase in Li metal battery, (c) the full battery performance comparison between conventional electrolyte and F-rich electrolyte, (d) the LUMO and HOMO energy comparison between EC, DMC and LiFSI.
此外,鋰金屬的利用率會嚴重影響鋰金屬的使用壽命,在鋰金屬電池實際應用中,設計低過量系數的鋰顯得尤為重要。Qiu等134設計了高濃度的三元體系的鋰鹽(LiTFSI-LiNO3-LiFSI)電解液以應用于提高鋰的利用率,其中,LiFSI和LiNO3能夠生成穩定的LiF-Li2O層,LiTFSI保證了高濃電解液的離子電導率和穩定性。在鋰過量系數低至0.5的條件下,使用此電解液的Li∥Cu和Li∥LFP電池分別能夠保持99.4%的庫倫效率和循環500圈后92.0%的容量保持率。一般來說,高濃電解液具有高氧化穩定性、高熱穩定性、低電壓波動、高離子遷移數等優勢,但是高濃電解液的價格較高且液體流動性差,因此在鋰金屬電池中需要設計合適鋰鹽成分和最佳濃度的電解液仍然具有挑戰性。
4.2 SEI穩定添加劑
在商業化鋰離子電池體系中,用于穩定石墨或者硅負極表面SEI的添加劑一直以來都受到廣泛研究和關注135,136,典型的成膜添加劑有FEC (氟代碳酸乙烯酯)137-139,VC (碳酸亞乙烯酯)140,141,ES (亞硫酸乙烯酯)142,143等。鋰負極表面的SEI膜受到枝晶生長的影響而更加的不穩定,因此需要探索合適的電解液添加劑穩定鋰負極。由于鋰的活潑性較高,金屬鋰基本會與所有鋰鹽和有機溶劑發生反應構成SEI,選擇成膜添加劑一般需要其在電解液組分之前和金屬鋰反應同時形成穩定的成分,因此好的成膜添加劑一般需要有以下幾種特性:i)添加劑有較低的LUMO能級和較高的HOMO能級從而能夠優先和金屬鋰反應;ii)添加劑和金屬鋰的反應產物需要保持好的化學和電化學穩定性,具有電子絕緣和離子導通的性質;iii)形成的SEI需要保持致密連續的結構。由于LiF具有較高的楊氏模量(~64.9 GPa)和電子絕緣性質(10?31S·cm?1)144-146,能夠很好滿足以上對反應產物的幾種需求,故而選擇合適的含氟添加劑在鋰金屬表面構筑富含LiF的SEI能夠有效地抑制鋰枝晶的生長。
Zhang等147在1.0 mol·L?1LiPF6-EC/DEC電解液體系中添加5% (w) FEC用于保護鋰金屬負極,如圖7a所示,由于FEC (?0.87 eV)相比于EC (?0.38 eV)和DEC (0 eV)具有更低的LUMO能級從而能夠優先和鋰金屬反應生成富含LiF的SEI從而使鋰離子能夠均勻沉積。研究表明,在電流密度為0.1 mA·cm?2,容量為0.5 mAh·cm?2的條件下添加5%(w) FEC的Li∥Cu電池體系庫倫效率可以在循環100圈后仍然維持在98%附近,而未添加FEC的電解液體系的庫侖效率僅維持在92%。當使用三元正極材料LiNi0.5Co0.2Mn0.3O2(NCM523)和金屬鋰組成全電池時,其中NCM523的負載量為12 mg·cm?2,添加5% (w) FEC的全電池能在電流密度為180 mA·g?1能夠穩定循環100圈,而未添加FEC的體系在50圈左右則發生斷崖式容量衰減,這種現象是因為鋰枝晶的不斷生長和死鋰聚集造成電池失效。
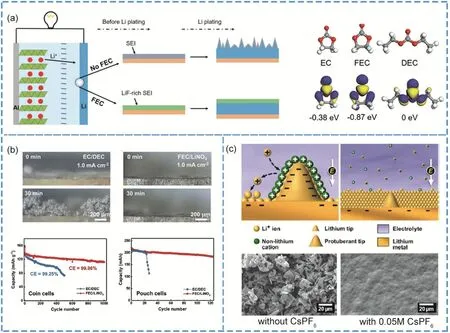
圖7 (a) FEC添加劑穩定鋰負極示意圖和LUMO能級對比147;(b) FEC和LiNO3協同保護鋰負極的斷面枝晶形貌和全電池性能比較153;(c) CsPF6誘導鋰沉積示意圖和鋰沉積形貌圖對比28Fig. 7 (a) Schematic of FEC additive protecting Li metal anode and the LUMO energy comparison between EC, FEC and DEC 147. (b) The sectional image of Li anode after cycling using conventional electrolyte and FEC/LiNO3 electrolyte, and the corresponding performance comparison full battery 153. (c) Schematic of inducing Li deposition via CsPF6 additive and the corresponding morphologies after cycling without and with CsPF6 28.
LiNO3常被應用于鋰硫電池體系中醚類電解液添加劑來抑制多硫化物的穿梭和鋰枝晶的生長148-150,LiNO3在醚類溶劑中很容易溶解,但是醚類溶劑體系由于其較窄的氧化還原窗口很難應用于高壓鋰金屬電池,所以對于高壓鋰金屬電池一般選用酯類溶劑體系,而LiNO3基本不溶于碳酸酯類溶劑中151。針對于這樣的問題,Yan等152探索了LiNO3在酯類電解液中的溶劑化化學問題,研究發現通過添加微量的CuF2能夠促進LiNO3在EC/DEC電解液體系中的溶解,添加LiNO3后的鋰金屬電池中Li負極表面生成含氮無機產物(Li3N和LiNxOy),能夠調節鋰的成核過程從而引導鋰的均勻沉積,在LFP和LiNi0.80Co0.15Al0.05O2全電池中相對于無LiNO3電解液體系展現出更穩定的全電池循環性能。Zhang等153結合了FEC和LiNO3的優勢將LiNO3溶解在DME電解液體系中然后再加入FEC構成醚/酯共溶劑化電解液體系解決了LiNO3不溶于FEC的問題。FEC和LiNO3共同參與形成Li+的溶劑化殼層并在鋰負極表面分解生成LiF,Li3N和LiNxOy等產物,這些產物構成穩定的鋰負極SEI并可以引導鋰離子的均勻沉積。如圖7b所示,當使用FEC/LiNO3體系時在1.0 mA·cm?2的電流密度下沉積半小時鋰金屬斷面結構仍然保持平整,而EC/DEC電解液體系生成大量枝晶鋰。在扣式和軟包電池中FEC/LiNO3電解液體系也展現出了更穩定的全電池循環性能。除以上添加劑外仍然有大量的含F、N、S、B等元素添加劑體系用于穩定SEI膜結構,如LiAsF6154,LiBOB (雙草酸硼酸鋰)155,LiDFOB (二氟草酸硼酸鋰)156,LiPS (多硫化鋰)157,H3BO3158等。
4.3 誘導鋰沉積添加劑
除上述和金屬鋰反應生成穩定SEI的成膜添加劑外,另一種類添加劑是通過電荷誘導調控鋰離子的沉積行為來抑制鋰枝晶的生長。如圖7c所示,Ding等28采用CsPF6作為添加劑來通過電荷誘導鋰沉積過程,根據能斯特方程可知當Cs+的濃度較低時,其還原電位會低于Li+,當負極外接電壓Va介于Li+和低濃度Cs+的還原電位中間時(即ELi+/Li>Va>ECs+/Cs),Cs+會被吸引在突起的鋰核表面,在帶電粒子相互作用下使其它的Li+沉積在非突起位置從而誘導Li+的均勻沉積。研究表明未添加CsPF6的鋰負極表面在沉積Li后生成了大量的鋰枝晶,而添加0.05 mol·L?1CsPF6的鋰負極在沉積Li后表面十分光滑平整,表明了CsPF6添加劑可以有效地調節Li+沉積行為。此通過電場自愈合策略也被進一步證明能夠有效地調控鋰沉積形貌159和提升鋰金屬電池性能160。
Ye等161報道了AlCl3作為電解液添加劑來抑制鋰枝晶的生長,AlCl3會和電解液中的痕量水反應生成Al(OH)3膠體顆粒,額外的Al3+吸附在顆粒表面使其帶正電,同樣地源于電荷誘導模型該帶電顆粒能夠調控鋰的沉積行為。Cheng等162使用了納米金剛石作為調控Li+沉積添加劑,通過DFT計算發現納米金剛石在(110)面具有最低的Li+擴散能壘并且表面對Li+具有更高的吸附能從而致使Li+可以吸附在納米金剛石顆粒表面,在鋰沉積的過程中以納米金剛石為成核位點均勻成核,研究結果展現了鋰沉積形貌呈致密的陣列狀而非枝晶狀,同時證明了具有高表面能和低擴散能壘的物質更易于誘導鋰的均勻成核和生長過程。
5 人造SEI改善鋰金屬負極
鋰金屬電池中鋰負極表面的SEI是活性鋰電極和有機電解液反應生成的一層鈍化膜,其主要成分由Li2CO3、LiOH、Li2O、LiF等無機層和ROCO2Li、ROLi等有機層構成。由于在電解液中原位生成的SEI成分復雜且不可控,不均勻的SEI結構和表面缺陷會導致鋰的不均勻沉積,鋰枝晶的生長會進一步破壞SEI結構,逐步增大電池極化和引發熱失控等安全問題164-166。因此在金屬鋰循環前于其表面構筑一層結構均勻穩定的,離子導通,電子絕緣的人造SEI是保護鋰負極的一種有效的策略166-169。由前面電解液部分討論可知LiF能夠鈍化SEI膜抑制鋰枝晶的生長,故而在金屬鋰表面設計穩定的富含LiF人造SEI能夠保護鋰負極。如圖8a所示,Zhao等170在350 °C條件下加熱含氟有機物CYTOP來產生氟氣(F2),然后F2和金屬鋰反應12h在鋰表面生成了一層厚度大約380 nm的LiF保護層。由該LiF保護的鋰負極構成的對稱電池在電流密度為1 mA·cm?2,脫鍍容量為1 mAh·cm?2的條件下能夠穩定循環300圈,并且循環后的鋰負極仍然保持穩定的形貌結構,無枝晶鋰產生。該氣固反應構筑LiF人造SEI的實驗方法簡單且能夠規模化制備,提供了保護鋰負極的新策略。在研究者們的進一步探索下,CuF2171、AlF3172、NiF2173、SbF3174、BF3·H2O175等含氟化合物用于在鋰負極表面構筑含LiF的保護層。Liang等176使用金屬氯化物MClx(M = In,Zn,Bi,As)的四氫呋喃溶液和金屬鋰先后反應生成LiCl和LixM合金層用于抑制枝晶鋰的形成。如圖8b所示,其中電子絕緣的LiCl可以阻止Li+在保護層表面還原成Li金屬,高離子電導的合金鋰化合物LixM能夠促進Li+快速遷移到合金層下在鋰表面還原成Li金屬,這兩方面的作用抑制了鋰枝晶的形成。在和Li4Ti5O12組成的全電池中,具有LixM/LiCl保護層的鋰負極相對于純鋰負極均表現出了更穩定的長循環性能。
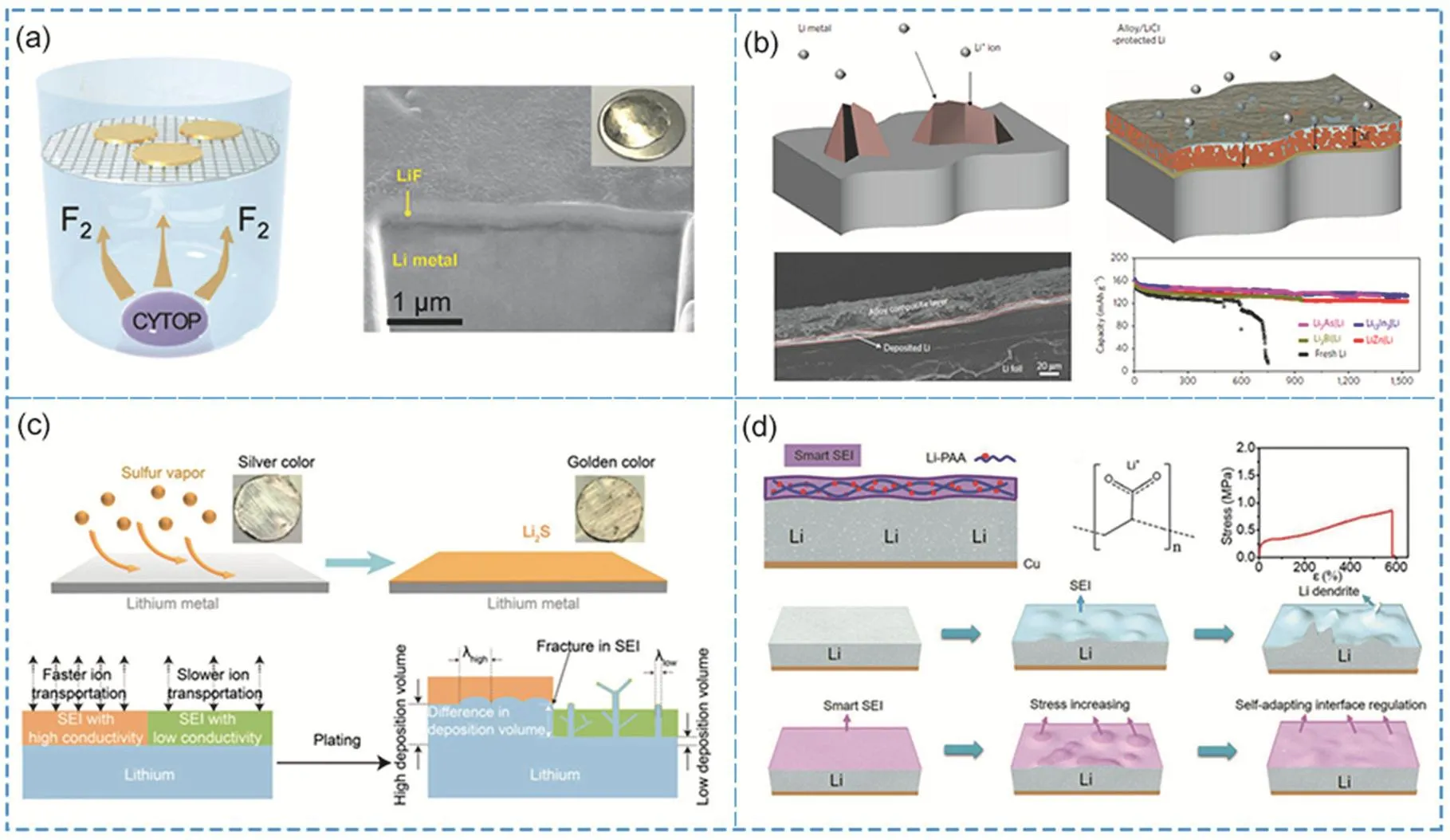
圖8 (a)氣相合成LiF保護層和形貌結構圖170;(b)金屬氯化物MClx (M=In, Zn, Bi, As)保護層的作用示意圖,斷面形貌結構和全電池性能對比176;(c) Li2S保護層合成示意圖及其作用機理183;(d) Li-PAA聚合物保護層保護鋰負極194Fig. 8 (a) Schematic of fabricating LiF protection layer and the corresponding morphology structure 170. (b) Schematic of Li deposition on the bare Li and MClx (M = In, Zn, Bi, As) protected Li, the corresponding sectional structure and full battery performance comparison 176. (c) Schematic of the fabrication process and effect of Li2S protection layer 183.(d) Schematic of Li-PAA protecting Li metal anode 194.
除第VII主族鹵化鋰具有較好的鋰負極保護作用外177-179,第VI主族元素和鋰形成的化合物也具有較好的離子電導率和鋰負極保護作用180-184。Chen等183在240 °C的條件下加熱單質硫產生氣體和金屬鋰反應24 h在鋰表面構筑高離子電導率Li2S (~10-5S·cm-1)無機保護層,合成過程和保護鋰負極作用如圖8c所示。作者通過COMSOL模擬計算得知當Li表面SEI的離子電導率越高時鋰離子流分布越均勻,不容易形成枝晶,而傳統SEI的鋰離子電導率一般都比較低,較慢的離子傳輸速率會引發鋰離子聚集而產生鋰枝晶。該Li2S保護的鋰負極構成的對稱電池在電流密度為1 mA·cm-2,脫鍍容量為1 mAh·cm-2的條件下能夠穩定循環750 h,極化電壓僅從初始的60 mV增長到90 mV。同樣Li2S@Li電極應用在LFP和LTO為正極的鋰金屬全電池中也保持更優的倍率性能和循環性能,而純鋰負極因枝晶生長問題而導致全電池容量呈斷崖式快速衰減。受到此Li2S設計策略啟發,Liu等184利用低沸點固溶化合物SeS2(~118 °C)作為氣體蒸發前驅體,將產生的硫化硒氣體和金屬鋰反應生成Li2S/Li2Se混合保護層,由于Se和S是同主族元素且半徑比S大,通過DFT計算得知Li2Se的離子遷移能壘比Li2S更低,故而能夠進一步提高Li2S體系的離子電導率,該Li2S/Li2Se混合保護層在對稱電池,LFP、NCM622、Li-S全電池中均取得了優異的電化學循環性能。除以上所述的無機保護層外,GeCl4185、MoS2186、Al2O3187,188、Li3PO4189、LixSi190、Mg191,192、Sn115,193等多種無機人造SEI均被廣泛研究于鋰金屬負極保護。
有機物聚合物由于其較高的韌性和機械穩定性也被作為穩定鋰負極固液界面層的研究對象。如圖8d所示,Li等194在金屬鋰表面構筑了彈性的PAA(聚丙烯酸)保護層,金屬鋰和聚丙烯酸原位反應生成聚丙烯酸鋰,即LiPAA,鋰離子的遷移即通過含Li聚合物鏈段遷移的方式進行。該LiPAA SEI層不僅阻止金屬鋰和電解液的副反應而且能夠自適應調節鋰枝晶對SEI層的應力,使金屬鋰能夠在此SEI層下均勻沉積。研究表明在對稱電池中,Li@LiPAA電極在電流密度為0.5 mA·cm?2,容量為1 mAh·cm?2的條件下能得到700 h的穩定脫鍍循環,并在LFP全電池中保持更穩定的循環性能。此外,應用于鋰負極人造SEI的聚合物還有PVDF-HFP(聚偏氟乙烯-六氟丙烯)195,高極化β-PVDF196,PDMS (聚二甲基硅氧烷)197,PEO (聚環氧乙烷)198等多種體系。
當單純有機聚合物體系的離子電導率無法進一步提高時,需要結合無機物和有機聚合物的優勢制備高離子電導率,高韌性的人造SEI層。Liu等199通過溶液法合成納米尺度的Cu3N顆粒,再將Cu3N顆粒和有機聚合物SBR (丁苯膠)溶于THF(四氫呋喃)構成均一溶液體系,然后將該混合溶液體系涂覆在鋰金屬表面,Cu3N納米顆粒會和金屬鋰原位反應形成高離子電導率的Li3N (~10?3-10?4S cm?1)顆粒,則此人造SEI兼聚高離子電導率和SBR聚合物韌性的特征,當Cu3N/SBR應用于Li∥Cu電池體系時,在電流密度為1 mA·cm?2,鍍鋰容量為1 mAh·cm?2的條件下,其酯類電解液體系中的鋰脫鍍循環能穩定進行100圈,并且庫倫效率維持在97.4%,相對于純鋰負極的快速衰減趨勢有很大的提升。此外,用于保護鋰負極的有機/無機混合保護層還有PEO/TiO2200,PMMA (聚甲基丙烯酸甲酯)/SiO2201,PEDOT-co-PEG (聚3,4-乙烯二氧噻吩單體-聚乙二醇)/AlF3202等。
不同的人造SEI策略及其電化學性能總結在表2中,可以看出人造SEI策略對鋰金屬的保護作用具有很大的提升。但是在高鋰利用率電池體系中,人造SEI的離子電導率和結構穩定性仍然有待改善,需要研究者們不斷的開發出優異的保護策略和探索其內在作用機理以進一步實現高比能量鋰金屬全電池。
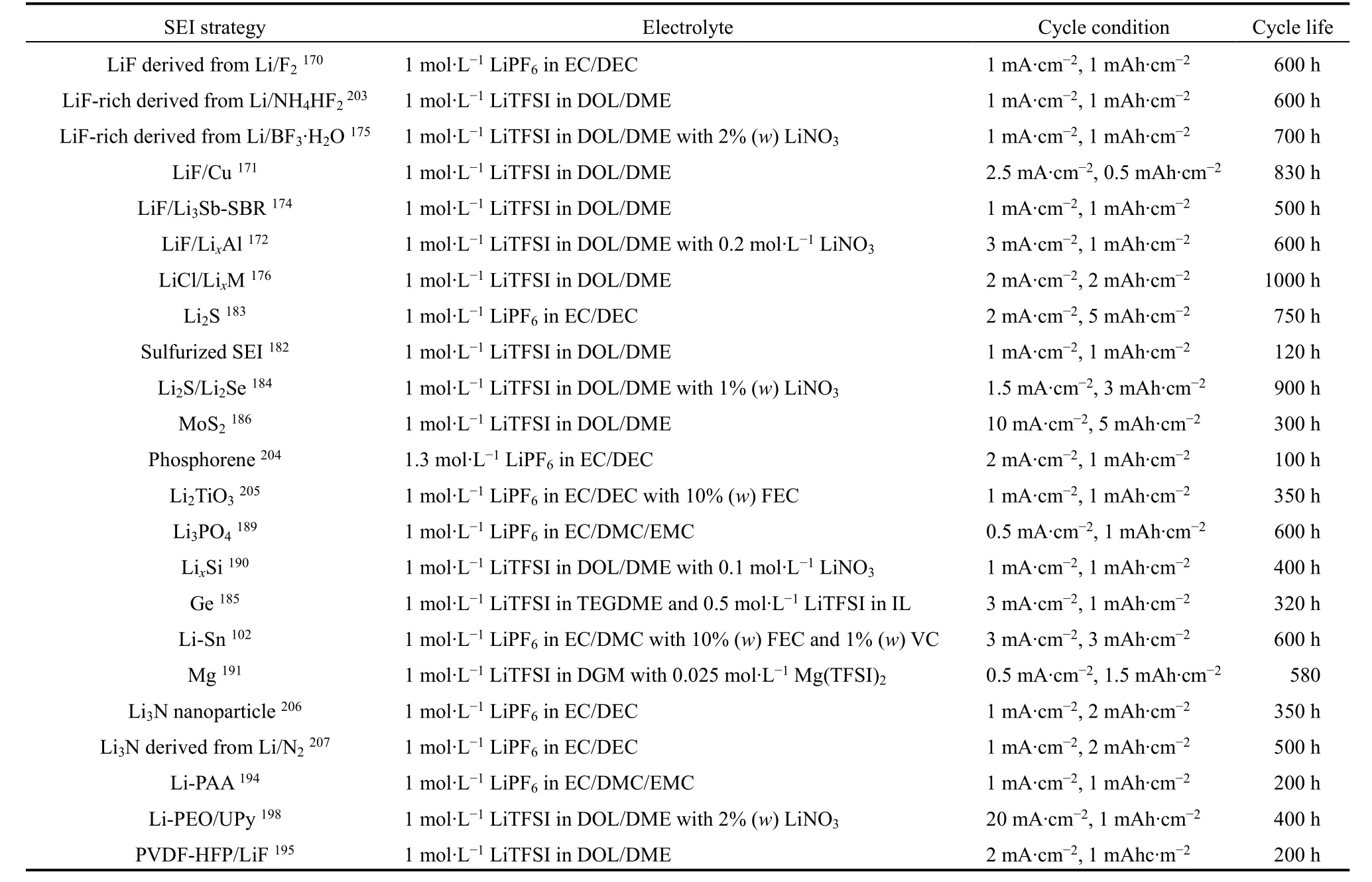
表2 不同 SEI策略的對稱電池性能對比Table 2 The cycle performance comparison of Li symmetrical cells with different artificial SEI.
6 修飾隔膜保護鋰金屬負極
鋰二次電池中隔膜的作用是阻隔正負極接觸避免發生短路現象,隔膜是允許溶劑化鋰離子穿過而不允許電子導通,在鋰金屬電池中,當鋰枝晶刺穿隔膜時則會連接正負極從而引發電池短路著火等安全事故。在鋰金屬電池中由于隔膜和鋰負極是處于緊密接觸狀態,因此,在商業隔膜上修飾無機或有機材料來提高隔膜抗枝晶應力和調控枝晶生長行為,并且相應的策略可以規模化擴大應用。Liu等208則通過在隔膜負極端表面修飾功能化碳層改變鋰枝晶的生長方向從而來抑制鋰枝晶的進一步生長。如圖9a所示,鋰枝晶的頂端電勢φt高于其底端鋰表面電勢φs209,且枝晶越尖銳頂端和表面電勢差越大,其電勢差是枝晶生長的驅動力,逐漸生長的鋰枝晶會刺穿隔膜并產生大量的死鋰。對苯磺酸(para-benzenesulfonic acid,縮寫為pb-SO3H)修飾的功能化納米碳層先被浸在LiNO3溶液中吸附一些鋰離子形成pb-SO3Li,然后在隔膜上涂覆此含鋰功能化碳層,如圖9b所示,由于功能化碳層和鋰金屬在電池中處于物理接觸狀態從而具有相同的電勢,在鋰金屬電池充電過程中鋰枝晶不僅從鋰表面生長也會從功能化碳層表面生長,當兩端枝晶頂端觸碰時,整體枝晶鋰的電勢差變為零從而失去鋰枝晶生長驅動力,后續的鋰則均勻沉積在碳層和鋰金屬層中間。研究結果展現出了在隔膜表面有一層均勻的鋰層而非枝晶鋰,并且當此功能化碳層應用在Li∥LFP全電池時,該全電池展現出了超過800圈的穩定循環和穩定的極化電壓值,而普通隔膜體系在220圈附近則出現了快速的容量衰減。
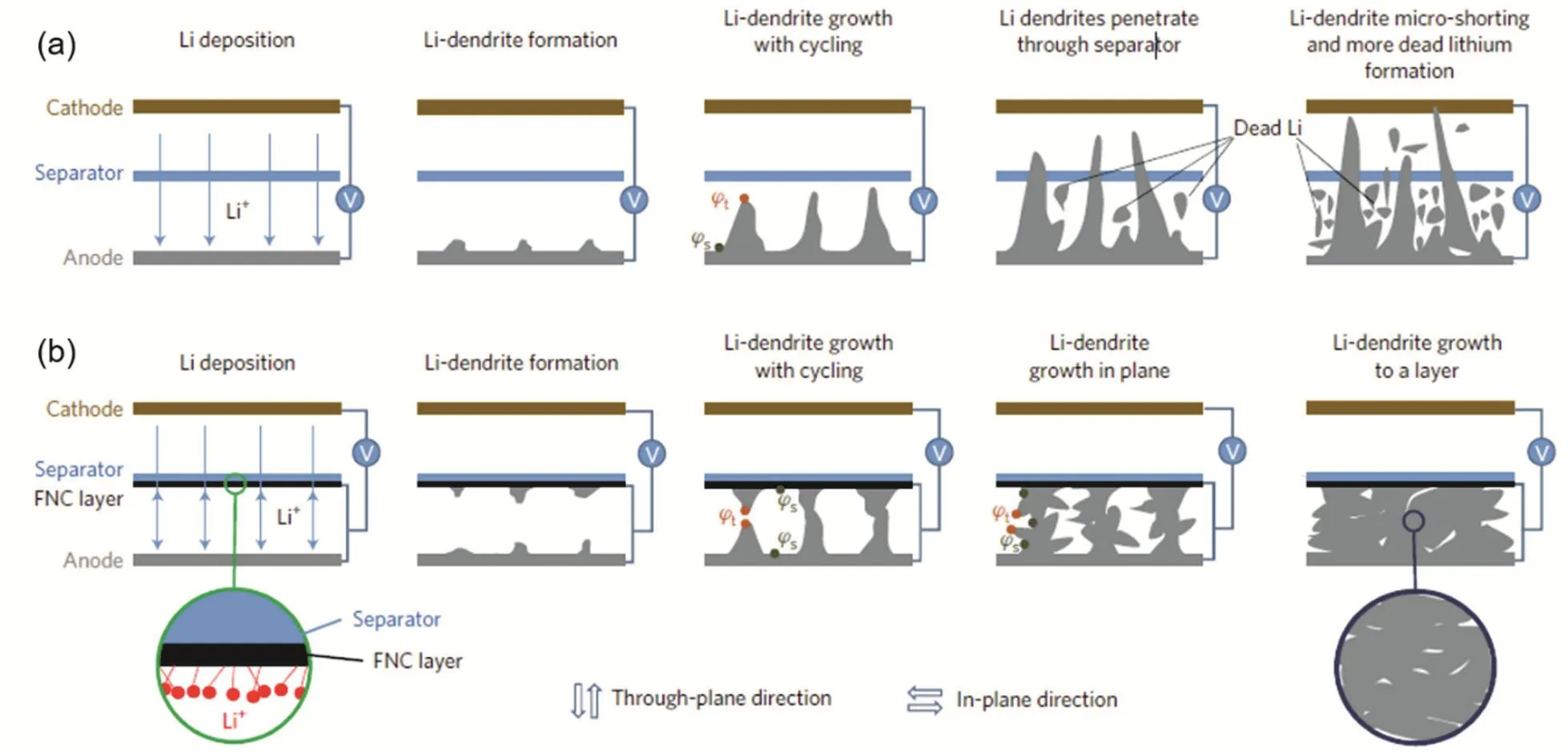
圖9 (a)傳統隔膜的鋰金屬電池體系中的鋰枝晶生長示意圖,(b)涂覆功能化碳層隔膜的鋰金屬電池體系中的鋰生長示意圖208Fig. 9 Schematic of the Li dendrite growth process in Li metal batter using normal separator (a)and FNC-coated separator (b) 208.
Li等210使用氧化石墨烯(GO)和聚丙烯酰胺(polyacrylamide)共聚構成多孔二維分子刷結構材料GO-g-PAM并將其涂覆在PP隔膜表面用于調控鋰離子的沉積,其中GO是分子刷材料的骨架,此二維結構能使鋰離子在電解液中快速傳輸,PAM中的C=O和N-H官能團能夠吸附鋰離子并使之在分子水平均勻分布,故而使鋰離子在集流體表面能夠均勻沉積,抑制了鋰枝晶的生長。研究表明當GO-g-PAM修飾的PP隔膜應用于Li∥Cu電池體系中,在電流密度為1 mA·cm?2,鍍鋰容量為1 mAh·cm?2的條件下能夠保持98%的庫倫效率穩定循環150圈,并且鋰金屬在循環后表面和斷面仍然保持穩定光滑的結構,未出現鋰枝晶和死鋰。此外,為了實現穩定的高能量密度鋰硫電池體系,BN211,MOF212,N-graphene213,PAN纖維214等修飾隔膜的無機材料不僅用于調控鋰離子的沉積,抑制鋰枝晶的生長,而且同時可以用于限制硫正極多硫化物的穿梭效應,從而實現了兼具安全性和穩定性的高能量密度鋰硫電池。同時,隔膜修飾保護鋰負極也會存在一些挑戰不可忽略,如隔膜厚度的增加會造成電池能量密度降低、鋰離子在溶液體系中擴散路徑變長、阻抗增大、極化電壓增高等,因此需要合理設計修飾層的厚度及其化學成分。
7 全固態鋰金屬電池
傳統鋰二次電池中使用的有機溶劑體系電解液是出現電池安全問題的根本原因,當將有機電解液替換為不可燃的固態電解質能夠從源頭上解決電池安全隱患215。但是使用固態電解質會造成電池體系能量密度降低,故而在全固態電池體系中使用高能量密度鋰金屬負極能進一步提高其能量密度,同時也解決了高活性鋰金屬存在的安全隱患,因此研究全固態鋰金屬電池(ASSLMBs)對于發展新型兼具高能量密度和安全性的電池體系具有十分重要的意義217。全固態鋰金屬電池一般存在以下幾種問題217:1)固態電解質的鋰離子電導率相對于有機電解液較低,造成高電流密度下離子傳輸受阻,極化增大;2)固態電解質和電極材料以及鋰金屬的固固界面接觸阻抗較大,導致界面處離子傳輸很慢;3)鋰金屬在全固態電池中會沿著電解質晶界生長鋰枝晶引發電池短路218,219。
固態電解質主要分為無機陶瓷固態電解質(ICEs)和有機聚合物固態電解質(SPEs)220,221。如圖10a所示,SPEs一般具有較好的柔韌性,和電極材料具有較低的接觸阻抗,但是強度低,能夠被鋰枝晶穿破;ICEs和電極材料接觸較差,但其硬度高,能夠阻止鋰枝晶的持續生長。結合二者的優點,Duan等222率先設計了非對稱的全固態電解質結構(ASE),以25 μm后的celgard隔膜作為電解質載體,在正極材料面設計厚度為5.4 μm的PEGMEA(聚乙二醇甲醚丙烯酸酯)聚合物電解質層,在鋰金屬面設計厚度為5.6 μm的致密Li7La3Zr2O12(LLZO)層,ASE的總厚度控制在36 μm,在ASE構成的鋰對稱電池能夠在0.1 mA·cm?2的電流密度下進行3200 h的脫離和鍍鋰循環,在Li/ASE/LFP全電池中穩定循環120圈,并保持99.8%的高庫倫效率。
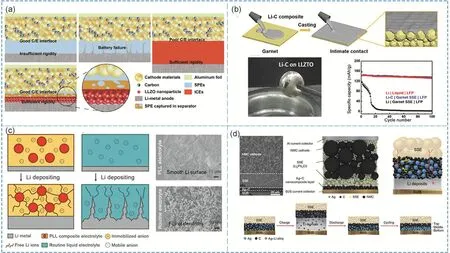
圖10 (a)使用聚合物電解質,無機陶瓷電解質和非對稱電解質的固態鋰金屬電池示意圖222,;(b)在LLZO固態電解質上涂覆Li-C復合負極示意圖和相應的光學照片以及全電池性能對比223;(c)在PLL固態電解質和液態電解液中鋰的沉積行為示意圖和相應的沉積鋰形貌圖224;(d)使用無鋰的Ag/C復合物作為負極的固態鋰金屬全電池結構圖和相應的示意圖227Fig. 10 (a) Schematic of solid Li metal battery using SPEs, ICEs and ASE as electrolyte 222. (b) Schematic of Li-C composite on LLZO electrolyte, the corresponding optical image and the full battery performance using LFP as cathode 223.(c) Schematic of Li deposition behavior and morphology structure in PLL solid electrolyte and liquid electrolyte 224.(d) Sectional structure and schematic of solid Li metal battery using Ag/C as Li-free anode 227.
然而,金屬鋰和固態電解質間的接觸阻抗也比較大,并且會生成鋰枝晶,當電流密度較大時,其高界面接觸阻抗則會嚴重增大電池極化,為了解決此類問題,Duan等223在熔融金屬鋰中加入適量石墨添加劑構成Li-C復合物,該熔融Li-C復合物展現出相對于純鋰更低的流動性和更高的粘度,如圖10b所示,然后將該熔融Li-C負極涂覆在石榴型無機陶瓷固態電解質Li6.5La3Zr1.5Ta0.5O12(LLZTO)上構筑均勻穩定的Li-C/LLZTO界面。研究發現LLZO對Li-C復合物具有更好的兼容性,熔融Li-C復合物能夠很好的吸附在LLZTO表面上,而熔融Li很難自發吸附在LLZTO上。通過斷面結構圖發現Li-C電極和LLZTO電解質緊密接觸,界面阻抗在11 ?·cm2,而Li電極和LLZTO存在幾個微米大小的縫隙,界面阻抗高至381 ?·cm2。得益于該穩定的界面,Li-C/LLZTO/Li-C對稱電池在0.3 mA·cm?2的條件下能夠穩定循環250 h,當Li-C/LLZTO和LFP組成全電池時,在0.5C的電流密度下可以和液態電解液體系中一樣穩定循環100圈,而使用純鋰負極時其全電池容量快速衰減至零。
為了在全固態電池體系中調控鋰/電解質界面上的鋰離子沉積行為,Zhao等224將Al摻雜的Li6.75La3Zr1.75Ta0.25O12(Al-LLZTO)的粉末加入到溶有鋰鹽的聚合物體系(PEO/LiTFSI)中并分散均勻,然后將該流延體涂覆在聚四氟乙烯板上并干燥制成均勻的聚合物-無機物固態電解質薄膜(PLL)。由于Al-LLZTO具有較高的離子電導率并且該顆粒的加入減小了PEO聚合物的結晶度從而使PLL在25 °C離子電導率高達1.12 × 10?5S·cm?1,并且其陽離子遷移數t+高達0.58,高于PEO/LiTFSI(t+= 0.37),1 mol·L?1LiPF6-EC/DEC (t+= 0.22),1 mol·L?1LiTFSI-DME (t+= 0.21)。如圖10c所示,其中TFSI?陰離子被聚合物基體和無機填充顆粒固定住,使界面處空間電荷和鋰離子均勻分布從而誘導鋰的均勻沉積,其鋰沉積形貌相對于液態體系中更加平整光滑,未出現枝晶狀的鋰。
近期,無鋰負極構成的鋰金屬電池受到廣泛的研究和關注225,226,此種電池結構需要正極的鋰能夠穩定均勻沉積在負極集流體上,并保持較高的鋰利用率才可以實現電池的穩定循環。如圖10d所示,Lee等227將60 nm的Ag顆粒和30 nm的導電碳黑粉末在Cu集流體上流延涂覆制成Ag/C層,然后使用Ag/C復合物作為無鋰負極,Li6PS5Cl為固態電解質,高鎳三元LiNi0.90Co0.05Mn0.05O2作為正極材料構件全固態電池。研究發現Ag/C層能夠有效地調節鋰沉積,當正極材料中的Li沉積到負極中時,鋰離子先與Ag形成合金Li9Ag4,然后再成核生長并均勻沉積,研究發現,部分的Ag顆粒會隨著鋰沉積過程遷移到集流體表面并維持在Ag/C層和集流體之間,在此Ag顆粒的誘導作用下,Li均勻沉積在Cu集流體和Ag/C層中間,當放電時,鋰也能夠均勻的脫出。當利用此全固態電池體系構建0.6 Ah容量的軟包電池時,在電流密度為0.5C(1.0C=6.8 mA·cm?2),溫度為60 °C的條件下穩定循環1000圈,并保持99.8%的庫倫效率,體現出此種體系全固態電池具有一定商業應用的化潛力。
不同界面策略構成的Li/電解質/Li對稱電池性能總結如表3所示,穩定鋰負極和電解質界面的策略主要通過以下兩方面實現:一是通過添加化合物飾鋰金屬電極來構筑快離子傳輸和低阻抗的Li/電解質的界面;二是修飾固態電解質以提高其親鋰性和調控鋰離子的沉積行為。但是固態鋰金屬電池在高電流密度下的應用仍然具有挑戰,需要進一步構建穩定的固固界面和開發出先進的電解質材料。
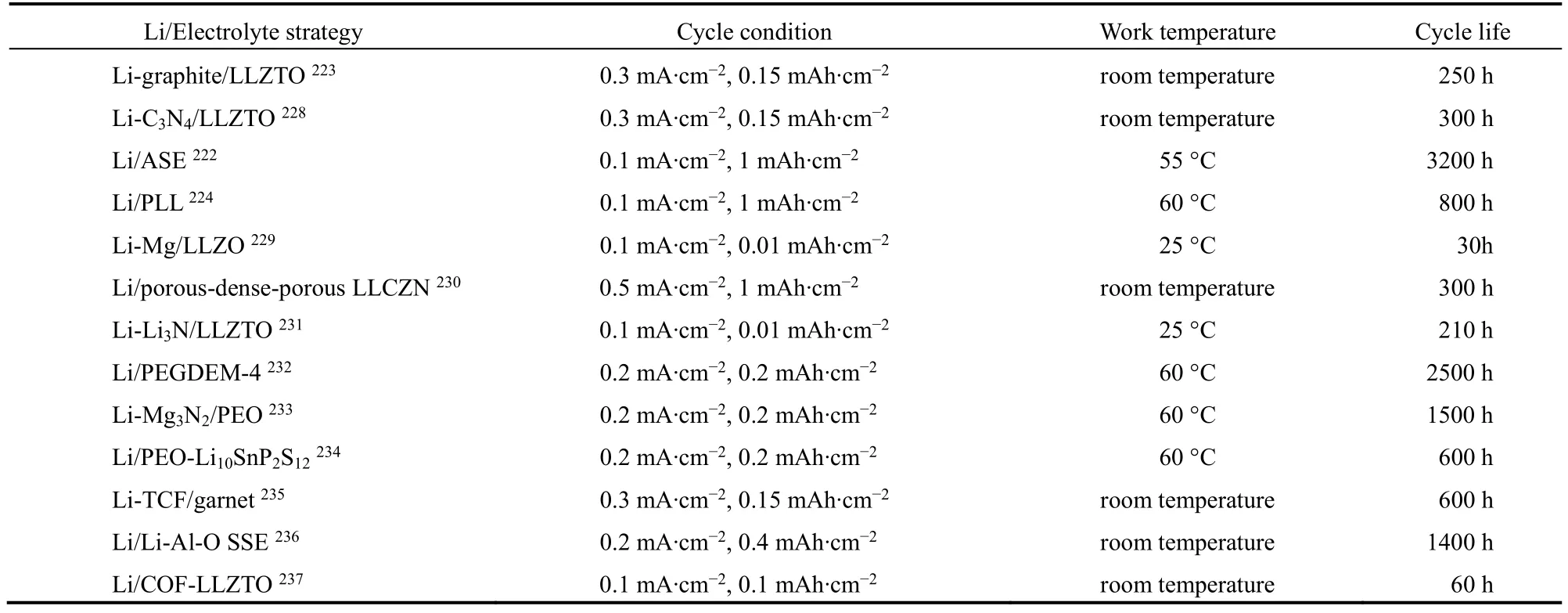
表3 固態鋰金屬對稱電池性能比較Table3 The cycle performance comparison of solid-state Li metal batteries.
8 總結與展望
鋰金屬負極材料近年來被視作提高電池能量密度的終極材料,然而金屬鋰的高活性和不可控的鋰枝晶仍然是引發電池安全隱患的重要原因,限制著鋰金屬電池的實際應用。過去幾十年里,鋰枝晶的成核和生長模型已經得到廣發的研究和深度的理解,其主要模型結構有表面聚集生長模型,擴散控制模型和電荷誘導模型等。導致鋰枝晶生長的主要有兩方面的因素,一是鋰的低表面能會使得鋰傾向于生長成一維枝晶,二是鋰離子在SEI層擴散速度較慢且離子流分布不均勻促使鋰不斷在突起的位置生長,故而鋰金屬負極在高電流低溫的條件下更容易生成枝晶。此外,鋰金屬的無限體積膨脹會導致死鋰的產生從而降低了電極的利用率。
基于鋰金屬的問題和挑戰,本文詳細總結了近年來針對于鋰金屬負極改善策略,主要包括三維儲鋰基體限和集流體,電解液和添加劑,修飾隔膜和人造SEI等。這些策略從鋰枝晶的生長原理和鋰表面SEI組分出發,主要功能在于限制鋰體積膨脹,降低離子流和表面電流密度,構建穩定和快離子傳輸的表面SEI。這些策略主要集中于實驗室規模,當鋰金屬電池要達到實際的應用條件時,電流密度和鍍鋰容量會進一步提高,則會帶來更多的安全問題。當鋰金屬匹配固態電解質時能夠不僅能夠從源頭上解決其安全問題,而且能夠提高電池體系比能量,但是全固態鋰金屬電池的界面高阻抗問題和較低的離子傳輸速率一定程度限制著其發展。鋰金屬負極目前仍然處于研究階段,在液態和固態體系均需要進一步探索鋰沉積行為機理,穩定的SEI和界面組分,開發出更先進的儲鋰基體骨架和電解質體系,推進鋰金屬電池的實際應用。
猜你喜歡
祝您健康·文摘版(2024年6期)2024-07-26 00:00:00
小讀者(2021年2期)2021-03-29 05:03:48
少兒美術(2020年3期)2020-12-06 07:32:54
現代裝飾(2020年11期)2020-11-27 01:47:48
中學生天地(A版)(2020年3期)2020-04-10 10:57:45
故事作文·高年級(2020年3期)2020-03-17 09:24:33
瘋狂英語·新悅讀(2019年11期)2019-12-18 05:14:16
華人時刊(2019年13期)2019-11-17 14:59:54
NBA特刊(2018年21期)2018-11-24 02:48:04
文苑(2018年22期)2018-11-19 02:54:14
