硫化氫深度吸附材料的研究進展
2021-03-06 02:58:02于濤王運東劉作華馬建修靖宇
化工學報 2021年2期
于濤,王運東,劉作華,馬建修,靖宇
(1 清華大學化學工程系,化學工程聯合國家重點實驗室,北京100084; 2 重慶大學化學化工學院,重慶400044;
3天津綠菱氣體有限公司,天津300457)
引 言
硫化氫(H2S)是一種無色、有臭雞蛋氣味的易燃有毒氣體,嗅覺閾值極低,其主要產生于煉焦爐,污水處理,食品加工工業,煤炭和天然氣、煉油工業[1]。硫化氫對人體危害較大,低濃度的硫化氫會引起發熱、頭暈、呼吸困難,高濃度的硫化氫則會導致窒息。在工業上,硫化氫的酸性和腐蝕性會對設備和管路造成嚴重腐蝕。同時,即使在濃度低至0.01%(體積分數,下同)的情況下,其也會導致燃料電池中的催化劑中毒失活[2]。因此如何將氣體中的硫化氫雜質清潔、高效、深度脫除已成為國內外學者的研究重點。
隨著社會的發展進步,越來越多的領域對工業產品的硫含量有了更高的要求。根據相關標準,半導體行業用氫含硫雜質(以H2S計)需低于10?7(體積分數,下同);低硫油氣產品的總硫含量不能超過10?5;而用于生產石墨電極的低硫焦硫含量需在5×10?6以下。近幾年發展迅速的新能源汽車車載燃料電池用氫的要求更高,為4×10?9[3]。基于以上要求,開發行之有效的深度脫硫技術迫在眉睫。
目前,H2S 的捕集脫除技術可分為干法和濕法兩大類。干法包括吸附法、膜分離法等;濕法包括醇胺溶液吸收法、離子液體法和生物脫硫[4?5]。濕法主要用于硫含量較高、規模較大的脫硫場合,而對于百萬分之一含量的深度脫硫則需要利用具有低能耗、高脫硫活性特點的干法進行。相較于普通脫硫,深度脫硫的操作濃度極低,反應的傳質推動力小,導致反應動力學低,反應程度不高。一般的吸附劑因其物理吸附弱和反應活性低而不適用于深度脫硫,因此,研制具有高硫容量、高選擇性、高熱穩定性、高脫硫活性和可循環再生的深度吸附材料是吸附法深度脫硫的核心,吸附材料的合成和改良要基于深度脫硫的特點進行。從20 世紀70 年代開始[6],碳基材料、多孔金屬氧化物等逐漸被用作吸附材料,隨著材料科學和納米技術的發展,沸石型分子篩、MOFs 成為國內外學者的研究熱點。本文以硫化氫的深度脫除為研究目標,綜述了碳基多孔材料、多孔金屬氧化物、沸石分子篩、MOFs 等材料用于脫硫過程的研究現狀,并對其發展提出建議和展望,以期為進一步設計和研制新型吸附材料提供借鑒。
1 碳基材料吸附劑
1.1 碳基材料吸附劑概述
碳基材料主要包括活性炭等多孔碳材料。活性炭是一種經特殊處理的炭,其大規模工業應用始于1991年[7]。將有機原料(果殼、煤、木材等)經過低溫炭化、活化可制得活性炭,圖1為其分子結構示意圖[8]。活化的過程是一個微觀過程,分子碳化物的表面被點狀侵蝕,使其具有無數細小孔隙,因此活性炭有較大的比表面積(500~1500 m2/g)和孔隙體積,這一特點是其被廣泛用作吸附材料的主要原因。

圖1 活性炭結構示意圖[8]Fig.1 Sketch of the atomic?level structure of activated carbon[8]
除活性炭外,其他多孔碳材料(如多孔碳球、多孔碳膜)以其巨大的表面積和高孔隙率引起了研究人員的興趣。盡管如此,由于多孔碳材料的孔隙一般為微孔,而且碳的疏水性不利于H2S 的吸附,因此,需改善多孔碳孔道結構,并通過浸漬、摻雜等手段進行修飾,以提高其吸附性能。
1.2 碳基材料吸附劑研究進展
活性炭對H2S 的吸附機理包括物理吸附和化學吸附。除了本身具有的吸附性外,活性炭能作為催化劑催化硫化氫的氧化反應。Wu 等[9]研究了使用Centaur20×50、WV?B、VA?507三種市售活性炭和以纖維素為前體制備的W?22 活性炭去除富氫氣體中的低濃度硫化氫。研究表明,活性炭的脫硫能力取決于其微觀結構和雜質,在適當的反應條件下,W?22 活性炭可將氣體中的硫化氫含量降低至十億分之一。此外,Shen 等[10]利用密度泛函理論(DFT)在分子水平對H2S 的吸附進行模擬,發現活性炭不僅為H2S的吸附提供活性位點,而且還能促進H2S分子的解離。H2S 解離后,與活性炭形成穩定的C—S、C—S—C和C—SH鍵,這與實驗數據相符。
活性炭的吸附性能受到其微觀結構和表面化學性質的高度影響。活性炭中的微孔和相對窄的孔徑分布能夠抑制氧化副產物SO2的生成,表面上氮基、氨基基團的存在能提高H2S 的穿透時間和穿透容量[11?12]。Seredych 等[13]以尿素(U)和三聚氰胺(M)為富氮前體,分別在450℃(A)和950℃(B)下對微孔活性炭S208C(S)進行摻雜改性,并用于沼氣脫硫。結果表明,氮基的引入為H2S 的解離提供了必要的堿度,在實驗條件下,吸附劑能在實驗開始后的580 min 內將出口氣體中H2S 濃度限制在10?4以下,H2S 的吸附容量可達71.4 mg/g。利用KOH、NaOH、K2CO3與Na2CO3進行化學浸漬的方法也常被用于活性炭表面改性[14?16]。堿浸漬改性能使商用活性炭吸附劑的吸附容量提高3~29 倍,且經過KOH和Na2CO3改性的吸附劑能將氣體中的H2S 降低至3×10?5。此外,Siriwardane 等[17]制備出納米MgO,并將其沉積于多孔活性炭表面制得改性活性炭。當納米MgO 的含量為1.2%(質量)時,改性活性炭的H2S吸附容量達275 mg/g,而原始的活性炭的吸附容量為53 mg/g,說明雖然MgO 堵塞了部分微孔,但其能夠通過化學吸附捕集H2S。DTG 分析表明,化學吸附機理包括H2S 的氧化和負載金屬的加成機制。Cimino 等[18]研究了浸漬ZnO 和CuO 的活性炭的吸附動力學,分析實驗數據得出兩種不同速率的H2S 氧化機制:其一是金屬氧化物團簇或表面氧物種的晶格氧迅速形成金屬硫酸鹽;其二是利用原料氣中水分和分子氧催化形成元素硫鏈。當O2和H2O 存在時,兩種機制共同起作用并獲得更好的H2S 捕集能力。當原料氣中含有高濃度二氧化碳時,金屬鹽浸漬活性炭表現出優于堿浸漬活性炭的脫硫能力。Cu、Cr 鹽浸漬的活性炭能夠將沼氣中的H2S 濃度從5×10?5降低至5×10?7以下,使其滿足熔融碳酸鹽燃料電池的要求[19]。活性炭吸附H2S 的相關研究結果總結如表1所示。
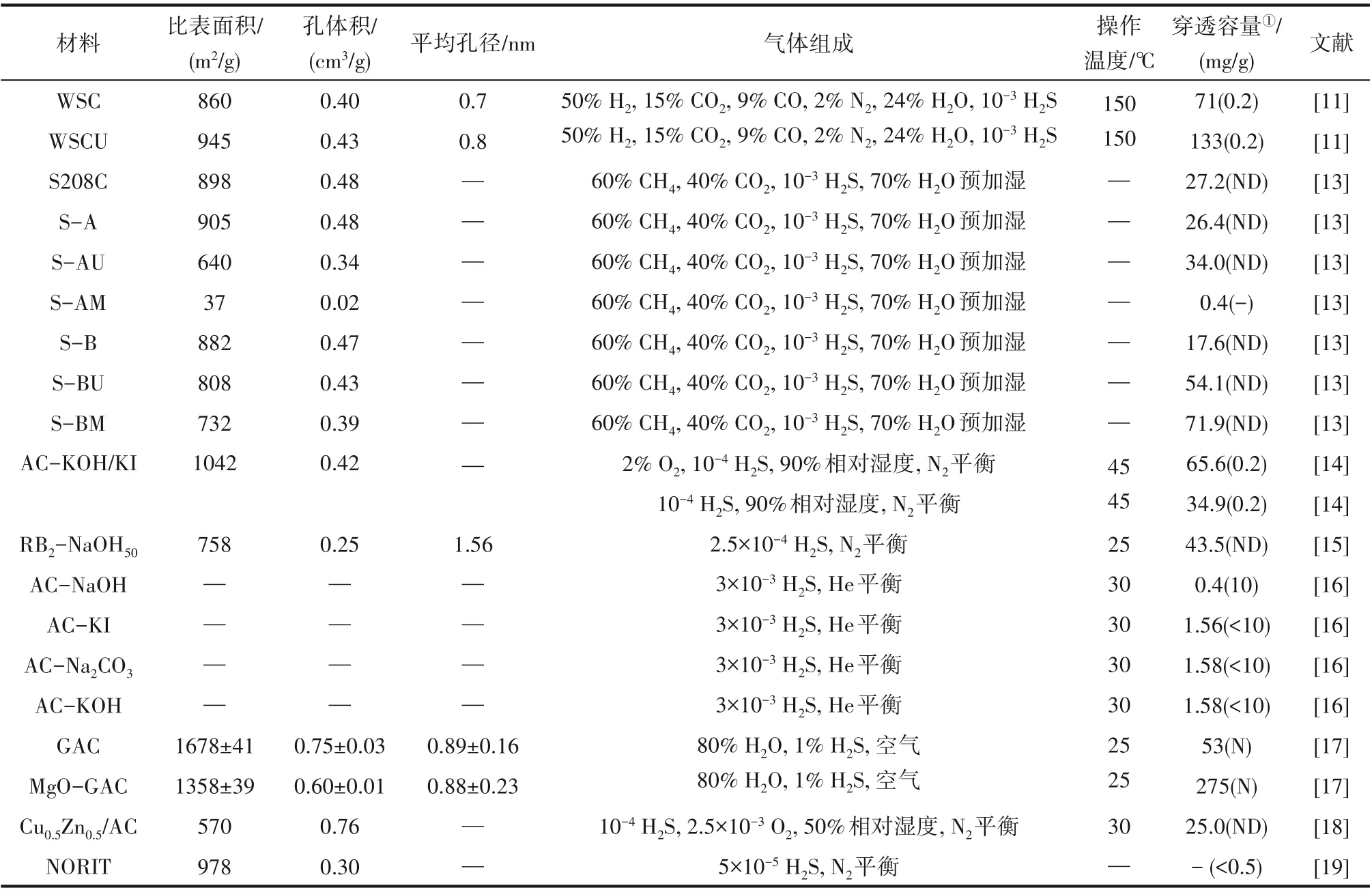
表1 不同活性炭的結構參數及H2S吸附性能Table 1 Parameters and adsorption ability of active carbon materials for H2S removal
其他多孔碳材料也常通過摻雜和浸漬的方法進行改性。當碳材料具有分級孔結構時,原子摻雜和分級孔結構對H2S 的吸附起協同作用:微孔能有效增加含有活性位點的面積,介孔則能減緩反應產生的硫堵塞孔道[20]。Qi 等[21]等以石墨質氮化碳(g?C3N4)為模板和氮源,通過相轉化法制備摻雜氮的多孔碳球(PCS),碳球直徑為1.2 mm,表面積為1017~1083 m2/g,具有微孔、介孔和大孔。將該碳球用于同時吸收混合氣體中苯和H2S,吸附過程如圖2 所示。結果表明,當氮含量為5.1%(質量)時,H2S吸附容量達40.1 mg/g,是原來多孔碳球的20 多倍。Zhang等[22]通過懸浮輔助納米塑形法制得千克級的無序介孔碳球(MCS),并分別用MgO、Na2CO3、NaOH、K2CO3與KOH 進行堿浸漬,脫硫實驗結果表明,相比于常規堿類,MgO 的浸漬效果更好。當MgO 含量為15%(質量)時,其H2S吸附容量最高,并且浸漬后MCS表現出光滑的外表面和均勻的MgO 顆粒分布,是一種極具潛力的吸附材料。相比之下,使用Na2CO3改性的單壁碳納米管(CNT)[23]在30℃下也能達到186 mg/g的H2S吸附容量,這不僅歸因于碳納米管獨特的空間結構為硫的儲存提供空間,還得益于Na2CO3增強CNT 的親水性和表面堿度,促進H2S 的吸附和解離。
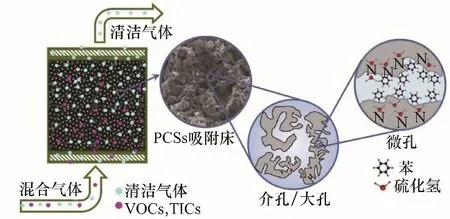
圖2 PCSs同時吸收苯和H2S過程示意圖[21]Fig.2 A scheme illustrating the simultaneous removal of benzene and H2S on PCSs[21]
總體來說,活性炭、碳纖維、碳納米管等碳基材料對H2S的高吸附容量和高吸附活性是基于發達的孔隙結構和表面官能原子、官能團而實現的。碳材料脫硫劑的失活是因為H2S氧化過程中產生的硫單質的沉積造成孔堵塞,因此,高介孔率材料以其較好的內部擴散動力學、產物硫易于擴散到外表面而具有更佳的脫硫能力。此外,吸附過程中的吸附動力學也尤為重要,如若吸附動力學不足,則吸附過程中的傳質速率低,單位體積吸附劑的脫硫能力低,所需的固定床體積增大,不利于大規模的工業生產。相較于活性炭,碳纖維和碳納米管具有更加完整、均一、穩定的結構,化學性能和可操作性更佳,在吸附領域具有很好的應用前景。目前,雖然已有多種碳材料能將氣體中的硫化氫脫除至百萬分之一以下,但這些大都是根據經驗來制備和改性的。為進一步提高碳基材料的吸附性能、節約空間和成本,對吸附過程的機理及吸附動力學的研究顯得尤為重要。
2 多孔金屬氧化物吸附劑
2.1 多孔金屬氧化物概述
1976年,Westmoreland等[6]利用最小自由能方法得出Fe、Zn、Mn、Cu 等11 種元素的氧化物具有高溫脫硫的潛力,金屬氧化物脫硫劑迅速成為研究熱點。經過幾十年的發展,金屬氧化物已被成功應用于催化、吸附領域,成為目前工業應用最為廣泛的催化劑。
金屬氧化物主要通過化學吸附脫除硫化氫,以ZnO吸附劑為例,反應機理[24?25]如下:
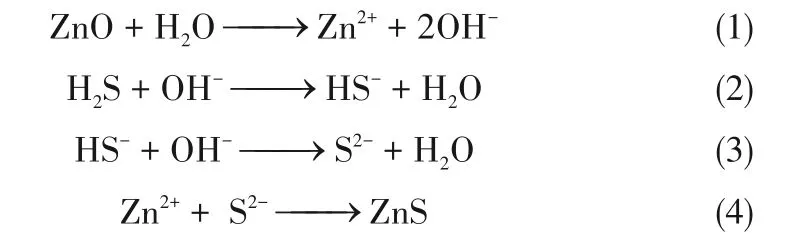
在脫硫過程中,H2S 經外擴散和內擴散并物理吸附到吸附劑表面或沉積到覆于其上的水膜中,然后與活性相(或氧化劑)反應生成硫化物(或硫酸鹽、單質硫),反應后氣體產物脫附并擴散至氣相主體。
基于以上機理分析易得,塊狀金屬氧化物因比表面積低、分散性差、孔隙率不足導致其難以用于脫硫過程,多孔金屬氧化物則能避免這些問題。相比于其他吸附劑,金屬氧化物脫硫一般在中高溫度下進行,較高的溫度使氣體的外擴散速率更快,吸附動力學更好;同時,在某些特殊有序結構(如3DOM)中引入金屬氧化物能進一步提高吸附過程中的內擴散速率,從而進一步提高吸附動力學。根據反應式和相關研究可以得出[26?28],水在脫硫過程中扮演重要的角色。一定量的水能夠促進反應的進行,過量的水則會使反應逆向移動,可能導致已經吸附的硫化氫的釋放。另外,為防止金屬氧化物脫硫劑的不可逆失活(生成金屬硫酸鹽),氣體中氧的含量也是至關重要的。因此,在未來的研究中,除開發具有開放大孔/介孔/分級孔以及添加助劑以獲得脫硫協同作用的新型多孔金屬脫硫劑之外,對于氣體組成、操作壓力、空速等條件的影響探究也是必不可少的。
2.2 多孔金屬氧化物吸附劑研究進展
近年來,針對Fe、Zn、Mn、Cu 等氧化物已有了大量的研究[29?30]。在這些金屬氧化物中,氧化鐵成本低廉,來源廣泛,且對H2S 的吸附具有較高的活性。Liu 等[31]以硝酸鐵為前體,制備出負載不同基團的介孔納米氧化鐵晶體,并使用高空速、高H2S含量的氣體研究了介孔納米鐵氧化物晶體的脫硫性能,發現硫容強烈依賴于吸附劑的孔體積和表面基團,孔徑范圍在2~4.5 nm 能得到更大的硫容量,羥基的存在也有助于H2S 的吸附。Long 等[32]通過水熱沉淀法制備出在室溫下吸附H2S 的低成本擠出型Fe2O3基吸附劑并進行脫硫實驗和數值模擬。脫硫結果表明,在不同條件下,脫硫率均達99%以上,吸附劑的吸附容量不低于16.0 mg/g(30℃±2℃,初始H2S 濃度為6×10?4~1.2×10?3)。MgO和TiO2能夠作為活性添加劑提高氧化鐵吸附劑的脫硫能力。當赤泥脫硫劑負載3%MgO 和10%TiO2時,其對熱煤氣的H2S 脫除率超過99%,原料氣中含有的水則會對硫容產生不利影響[26]。此外,將鐵摻雜到氧化銅中制備出鐵銅復合吸附劑能在40℃下將CO2氣流中的H2S 從10?3脫除至10?7以下[33]。
與多孔氧化鐵相比,多孔氧化鋅具有更高的脫硫精度和脫硫穩定性。不同的配方和制備技術會對ZnO 基吸附劑的脫硫性能產生很大影響。Tran[34]以瓊脂糖凝膠為模板分別制備高度多孔的ZnO 和摻雜Ni 的ZnO 吸附劑,并在400℃下對H2S 含量為4×10?4的氣體進行脫硫,摻雜Ni 的ZnO 吸附劑能將H2S含量降低至10?5以下,且穿透時間達市售ZnO 的五倍。硫容方面,多孔ZnO 的H2S 吸附容量為457 mg/g,是市售ZnO 吸附劑的兩倍;而當多孔ZnO 中含有4%(質量)的Ni 時,吸附容量進一步提升至730 mg/g,并且負載硫的吸附劑能通過熱再生完全恢復脫硫能力。脫硫能力的增強一方面在于合成的ZnO具有極高的孔隙度和相互連接的大孔/中孔(圖3),另一方面在于Ni 的摻雜為吸附反應提供了額外的活性位點。Zheng等[35]利用半焦和硝酸鋅,通過高壓浸漬法制備氧化鋅基吸附劑。高壓浸漬法能將半焦的表面積由16.65 m2/g 擴大至272.59 m2/g,并將納米ZnO 均勻分散其上。在300~550℃之間,浸漬20%ZnO 的吸附劑可將模擬煤氣(H2S 含量為3×10?4)中的H2S濃度降低至10?7,穿透時間達23 h。利用同樣的方法制得的Zn?Mn?Cu 基吸附劑能將CO和H2的混合氣體流(H2S 含量5×10?4)脫硫至10?7,并保持56 h[36]。另有研究表明[27],將ZnO用碳酸銨溶液預處理,與黏合劑混合后擠出所得的ZnO 基吸附劑具有良好的H2S 脫除能力,其對含有8×10?6的H2S 的混合氣體進行脫硫時,H2S 出口濃度低至2×10?8,即使在20 h 后,該數據僅為3×10?7。對于實際含硫氣體,除無機硫外,其通常還含有少量有機硫,如羰基硫(COS)、硫醇等。ZnO吸附劑雖對H2S有較強的吸附能力,但對COS 的吸附能力較弱。當氣體中存在COS 時,ZnO 的吸附效率會出現明顯下降[37]。Yang等[28]使用溶膠?凝膠法制備出異質結構三元復合吸附劑ZnO?Co3O4/SiO2,并進行穿透實驗。與Co 摻雜ZnO/SiO2和Zn摻雜Co3O4/SiO2相比,異質結構吸附劑的性能明顯增強,H2S 吸附容量最高達170.8 mg/g。重要的是,用完后的異質結構吸附劑能直接用作COS 的氫化催化劑,其在200℃下能使COS 完全轉化,遠高于商用Co?Mo/Al2O3催化劑的轉化率(30%)。

圖3 市售ZnO(a)、多孔ZnO(b)與Ni摻雜多孔ZnO(c)的SEM圖(圖中標尺相同)[34]Fig.3 SEM images of commercial ZnO(a),porous ZnO(b)and Ni?doped ZnO(c)(The scale bar is the same for all images)[34]
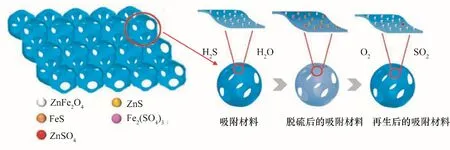
圖4 3DOM?ZnFe2O4/SiO2脫硫/再生示意圖[38]Fig.4 Scheme of sulfurization/desulfurization on 3DOM zinc ferrite composited silica sorbent[38]
金屬氧化物的脫硫過程是一個典型的氣固非催化反應,反應首先發生在表面,而后擴散到內部本體。在高溫脫硫時,氣體內擴散成為反應的控制步驟[38]。三維大孔結構(3DOM)具有高度有序的且相互連通的大孔,其能大大增強氣態被吸附物從吸附劑表面到內部活性位點的擴散[39]。將該結構引入H2S 吸附劑能顯著提高吸附反應動力學。Fan 等[25]通過膠體晶體模板法制備出的Fe2O3和Fe2O3/SiO2基3DOM 吸附劑能將模擬氣中的H2S 含量從3×10?4降低至2×10?7以下,脫硫率接近100%,反應速率提升一倍。Wang 等[40]以同樣的方法制備出具有3DOM結構的ZnO?SiO2復合材料,并在室溫下脫除H2S。SiO2能使ZnO 分散均勻并維持孔的完整性和強度。脫硫實驗結束后,將吸附劑置于500℃的空氣流(流速為100 ml/min)中進行熱再生,加熱時間4 h。在四次脫硫循環后,硫容量仍能達到初始狀態的67.4%,該數據還可以通過改變再生條件(溫度、氣氛組成)繼續提高。不同金屬氧化物的組合也能對H2S 的吸附產生協同作用,具有3DOM 結構的ZnFe2O4?SiO2吸附材料的脫硫性能優于單一氧化鐵或氧化鋅,當ZnFe2O4含量為70%時,能將實驗氣體中的H2S 含量從0.1%(體積分數)降低至5×10?7以下,且能獲得最大的H2S 吸附容量[38],脫硫和再生過程會有硫酸鹽[Fe2(SO4)3和ZnSO4]的產生(圖4)。這些結果說明具有3DOM結構的吸附劑具有很大的脫硫潛力。
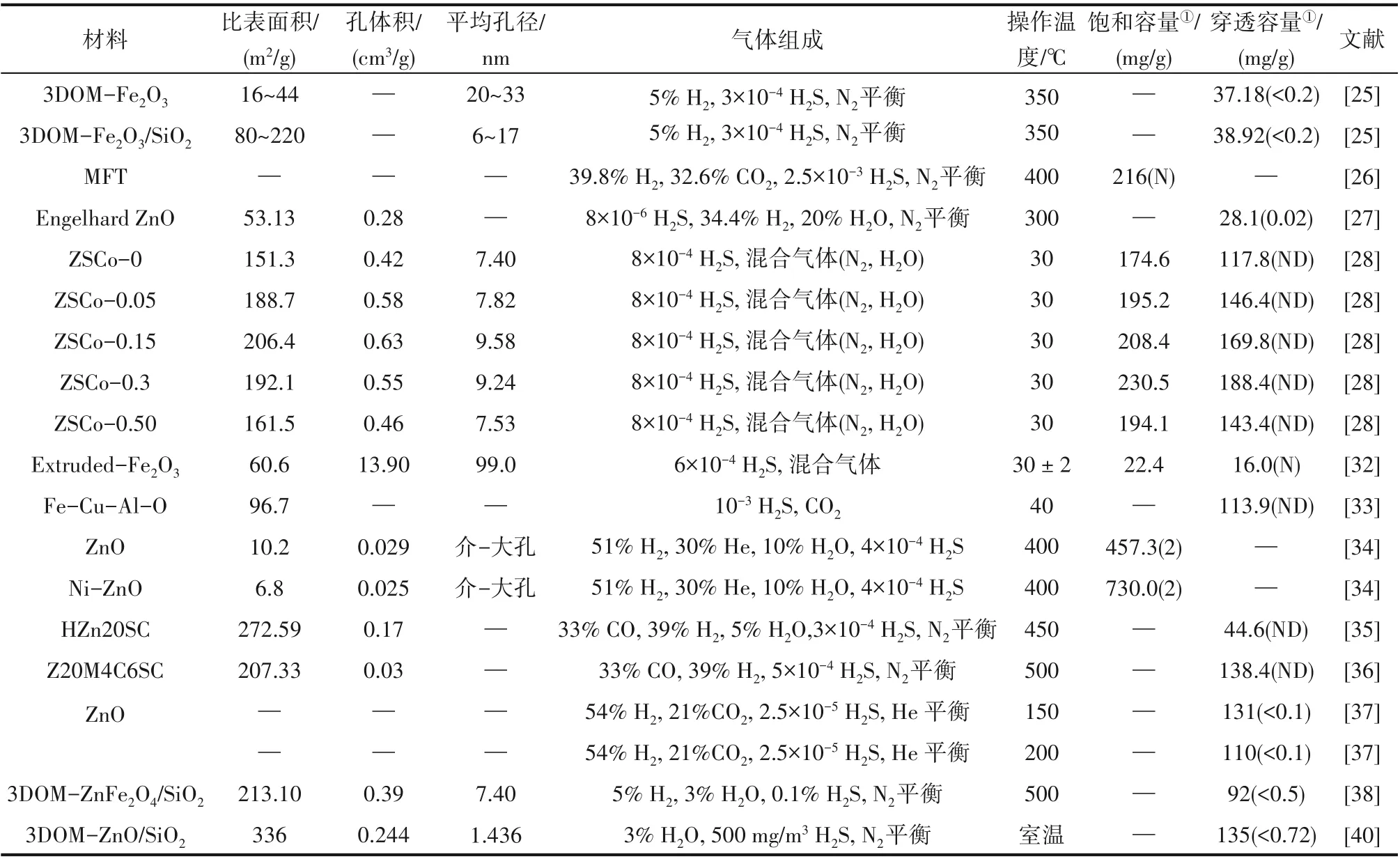
表2 不同多孔金屬氧化物的結構參數及H2S吸附性能Table 2 Parameters and adsorption ability of porous metal oxide materials for H2S removal
多孔金屬氧化物吸附H2S的相關研究結果總結如表2所示。
3 沸石分子篩吸附劑
3.1 沸石分子篩概述
沸石是結晶型的多孔鋁硅酸鹽,化學式可表示為Mx/n[(AlO2)(SiO2) ]·wH2O。沸石分子篩的骨架由和三維四面體構成,通過共用所有的氧原子相互連接[41],具有規則的晶內腔和分子孔道。
鋁硅酸鹽結構帶負電荷,陽離子(金屬離子、H+等)的存在使整體骨架保持電中性[42]。沸石分子篩的吸附適用性取決于Si/Al比、孔徑大小、極性、拓撲結構、陽離子種類等因素[8]。通常來說,高硅沸石具有較少的結構缺陷,更好的疏水性和水熱穩定性[43],適用于對非極性化合物的吸附。
沸石作為H2S 吸附劑具有成本低、可用于室溫脫硫等優點,吸附脫硫機理主要有兩種:硫化物與吸附劑的π 絡合和形成硫?金屬(S?M)化學鍵[24]。然而,相比于其他類型的吸附劑,原始沸石的脫硫效果并不理想,需對其進行改性和修飾。其中,金屬和金屬氧化物受到研究人員的青睞,這些活性物質的加入能改善沸石的吸附活性和硫化熱力學。雖然金屬改性的沸石脫硫劑的研究已取得一定的進展,但對于沸石微觀結構調控和吸附反應動力學的研究略顯不足,這些方面將是未來沸石吸附劑的研究重點。
3.2 沸石分子篩吸附劑研究進展
迄今為止,已發現大約五十種天然沸石,其中,斜發沸石(HEU)的研究最多,因其具有較低的酸性中心,相對高的堿性中心,被認為可作為深度脫硫的吸附劑[8]。相比于合成沸石,天然沸石雜質含量多,結構缺陷更加明顯,必須加以修飾才能得到更好的吸附性能。Alonso?Vicario 等[44]將天然斜發沸石在一定條件下活化,并與5A 型分子篩、13X 型分子篩做對比,發現活化后的天然斜發沸石具有更強的H2S 脫除能力和更大的吸附容量,能將模擬沼氣中的H2S 從0.1%降低至3×10?6以下,并且經過多次吸附?解吸循環后可以在氮氣氣氛下完全再生,再生溫度為280℃。合成沸石的研究也受到了廣泛的關注。Liu 等[45]由凹凸棒土(硅源)在不同條件下合成用于H2S 脫除的4A 型沸石分子篩,在最佳合成條件(硅鋁比和鈉硅比均為1.5,水鈉比為30,結晶溫度90℃)下合成的沸石的穿透容量和飽和容量分別為10 和15 mg/g,并且H2S 去除率接近100%。吸附過程的動力學符合Bangham 模型,吸附速率受到孔內擴散的限制。然而,對于其他組分競爭性吸附以及雜質對吸附的影響作用尚不明確。Yang 等[46]在水蒸氣存在的條件下研究了13X型沸石分子篩對克勞斯尾氣的脫硫性能,發現13X 對H2S和SO2的脫除機理是吸附?氧化還原過程,而13X 中的晶面(111)和(220)是主要的活性中心,其可在5.5 h 內將尾氣中的H2S 濃度限制在10?5以下。但在脫硫過程中,H2S 被氧化為單質硫,SO2被氧化為硫酸附著在沸石上,致使其再生性有所欠缺。
為進一步增強沸石分子篩的吸附能力,提高吸附劑對H2S 的吸附量和選擇性,需要對分子篩進行改性。沸石結構中的大空位則給大離子/大基團的引入提供了條件。Chen 等[47]使用離子交換法制備了Zn、Co和Ag改性的NaX 分子篩,其微觀形貌如圖5所示。H2S與金屬離子在吸附過程中形成S?M 鍵,而S?Ag鍵具有最強的相互作用,故AgX沸石表現出了較好的脫硫性能,H2S 的穿透容量為48.96 mg/g,穿透時間接近5 h,并能在350℃的空氣氣氛中加熱5 h 循環再生。與之類似,經過Cu 離子改性的13X型沸石分子篩對百萬分之一含量H2S的吸附能力顯著增強[48],能將沼氣脫硫至5×10?7以下,并用于熔融碳酸鹽燃料電池系統原料氣脫硫,同時具有再生性好、風險低的優勢。除此之外,Cu、Zn、TiO2、ZnO等[49?51]浸漬的方法也常被用于X 型和Y 型沸石分子篩的改性,改性后分子篩的H2S吸附能力顯著增強,比改性前提高3~30 倍。改性沸石分子篩對H2S 的吸附機理也通過DFT 得到論證[47,52]。沸石分子篩吸附H2S的相關研究結果總結如表3所示。
4 MOF材料吸附劑
4.1 MOF概述
金屬?有機骨架化合物(MOF)又稱多孔配位化合物,首次提出于1995 年[53],是一種新型多孔材料。MOF 是由金屬簇與有機體橋聯配體通過配位作用自組裝形成的,具有三個重要的組成部分,即金屬簇、有機配體和孔道。這三個組分可以作為結構基元構成一個基本的功能單元,功能單元在三維空間內的周期性拓展形成MOF材料。

圖5 NaX、CoX、ZnX和AgX的SEM圖[47]Fig.5 SEM images of NaX,CoX,ZnX and AgX[47]
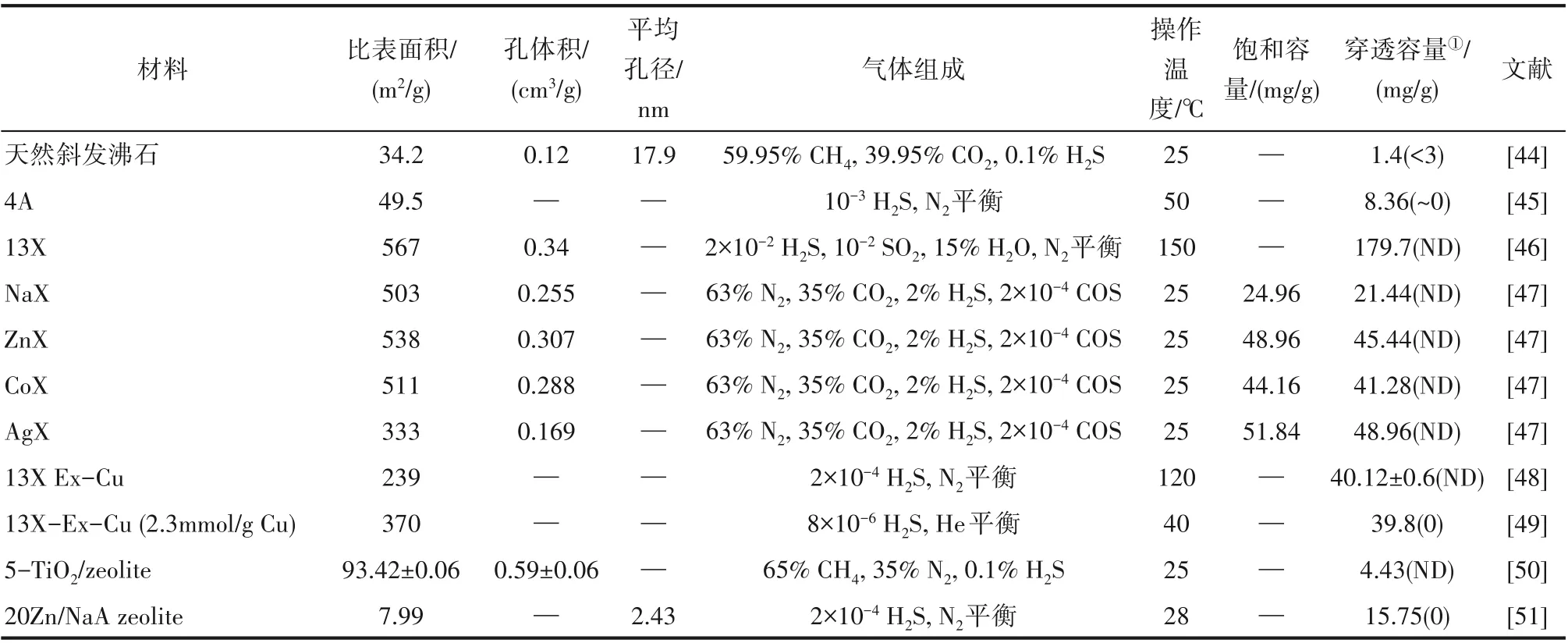
表3 不同沸石分子篩的結構參數及H2S吸附性能Table 3 Parameters and adsorption ability of zeolite materials for H2S removal
目前,有20000 多種MOF 被開發和研究[54],因MOF 材料具有高比表面積(1000~7000 m2/g)、結構豐富可調、金屬離子和配體種類豐富、具有不飽和金屬位點等優良的特點[55],故其成為理想的氣體吸附材料。然而,將MOF 用作H2S 吸附劑的研究還相對較少。已有的研究表明,雖然MOF 的高孔隙率和高表面積使其適用于氣體的吸附分離,但原始MOF的吸附容量和吸附動力學較差,必須進行活化和修飾。其中,利用摻雜金屬離子、活性基團等手段對MOF 進行表面改性是被證明的較為行之有效的方法,在實驗室條件下能夠將含硫氣體中的H2S 含量降低至10?6;將MOF 與其他多孔材料以及其他分離單元相結合制備新型復合材料是MOF 吸附劑的另一大發展方向,MOF 復合材料能協同發揮不同材料的吸附性能,提高氣體選擇性和吸附容量,但其實際應用還需進一步的研究。
4.2 MOF材料吸附劑研究進展
MOF 材料具有大比表面積,超高孔隙率,良好的可修飾性和開放的金屬位點。MOF對H2S的吸附研 究 始 于2009 年,Hamon 等[56]對 比 了6 種MIL 型MOF 材料的吸附等溫線和再生性能,發現雖然大孔MOF 較小孔MOF 有更高的H2S 吸附能力,但是吸附劑再生性變差。吸附劑的再生性能依賴于吸附前后MOF 骨架被破壞的程度。相比于H2S 吸附前,吸附后的MIL?47、MIL?53(Al)和MIL?53(Cr)的骨架結構幾乎不變,而大孔MIL?100 和MIL?101 的結構分別出現10%和40%的坍塌。而在另一項對MIL 的研究中[57],作者發現對于含有較低H2S 分壓的工業氣體流(如沼氣流),大孔MIL?101 能夠對H2S 進行可逆吸附,吸附機理基于H2S 分子與MIL?101 中不飽和位點的相互作用。而當混合氣體流中含有CO2時,介孔MIL?101(Cr)對H2S 的選擇性要優于UiO?66(Zr)和MIL?125(Ti)[58]。MOF 材料吸附H2S 的相關研究結果總結如表4所示。
許多MOF 材料都具有開放的不飽和金屬配位,但這些配位通常在合成過程中被溶劑分子或空氣中的小分子占據,導致原始MOF 的吸附能力有限。活化處理則可以釋放這些位點,清除通道,為有效吸附特定的客體分子留下空間。Li等[59]通過預熱處理將MOF?199 進行活化,脫硫結果表明,當活化溫度為180℃時,MOF?199的H2S穿透時間和穿透容量隨著吸附溫度的增加而增加,最高分別達4 h、57.2 mg/g。將胺類直接接枝到MOF?199 的金屬中心也能提高其H2S 吸附性能[60],其中,三乙醇胺(TEA)的效果最佳,H2S 穿透時間超過6 h。吸附能力的提高是因為吸附結合能的提高,胺類中的羥基與H2S 形成了牢固的OH—S 鍵。將金屬離子整合到MOF 結構中能夠提高其脫硫能力。Nikerl 等[61]使用不同的金屬鹽溶液對UiO?67(bipy)進行官能化,如圖6 所示。當原料氣中H2S 含量為10?3時,負載銅鹽的UiO?67(bipy)顯示出最好的脫硫性能,并且硝酸根、硫酸根和乙酰丙酮鹽對骨架具有穩定作用,而負載Cl?的骨架在吸附H2S時會塌陷。這些結果表明使用金屬鹽對UiO?67 官能化在百萬分之一含量H2S 脫除方面具有應用潛力。
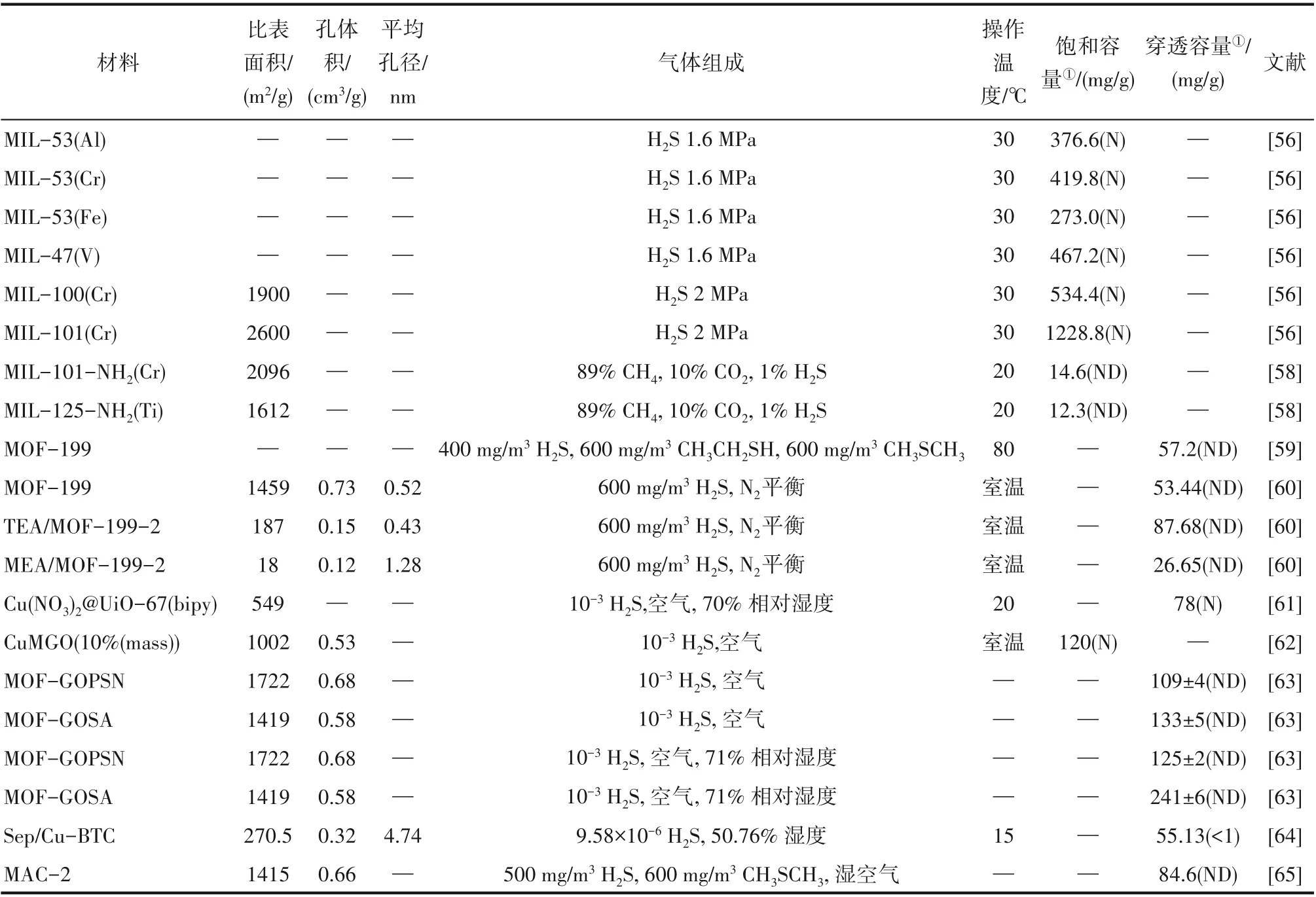
表4 不同MOF的結構參數及H2S吸附性能Table 4 Parameters and adsorption ability of metal organic frames for H2S removal

圖6 將金屬鹽整合進入UiO?67(bipy)示意圖[61](綠色八面體代表[Zr6O4(OH)4]12+團簇;灰色、紅色和藍色球體分別代表碳、氧和氮原子;橙色球體代表不同的金屬復合體)Fig.6 Postsynthetic insertion of metal salts into UiO?67(bipy)[61](Green octahedra represents the[Zr6O4(OH)4]12+cluster.Grey,red and blue spheres represent carbon,oxygen and nitrogen atoms,respectively.The orange spheres represent the respective metal complex)
除了對MOF 材料進行改性以外,利用MOF 制備復合材料吸附劑的相關研究也在進行。Petit等[62]將MOF 與氧化石墨(GO)合成雜化材料Cu?BTC/GO、MOF?5/GO 等,發現MOF 和GO 對H2S 吸附具有協同作用,合成材料對H2S 的吸附能力相比母體材料有了明顯提高。而Cu?BTC 和摻雜S、N 的GO 進行合成時,該材料的表面積提高了近一倍,具有良好的空隙率且磺酸基和胺基等極性反應性基團的引入提高了表面活性,H2S 主要通過酸堿反應以銅/硫鹽的形式被捕集。當實驗氣體含有10?3的H2S時,在潮濕條件下,負載磺酸基的復合材料的H2S 穿透時間約為5.7 h,且在前4.2 h 內,出口氣體中的H2S濃度接近0[63]。另一種含銅MOF材料Cu?BDC(對苯二甲酸酯配體)能將氣體流中的H2S濃度從10?5降低至10?6以下[64],但穿透時間相對較低,為0.76 h,且穿透時間隨著水分的增加而減少。將活性炭與MOF材料結合也能提高材料的H2S 吸附性能,Fan 等[65]發現當MOF?199 復合材料中含有2%的活性炭時,其在保持原來的微觀形態的情況下表現出了更有序的結晶結構和更大的表面積,最高硫容量比原來增加51%,當氣體中含有500 mg/m3的H2S 時,穿透時間超過6 h,但由于該過程的機理是化學吸附,生成的CuS 會破壞材料的結構。此外,為了進一步提高H2S 吸附過程的經濟性和選擇性,MOF 負載離子液體[66]、MOF 與膜分離[67]整合的研究也在進行中,初步結果表明這些新材料在氣體深度脫硫方面具有很大的應用潛力。
5 總結與展望
由于H2S 的毒性和腐蝕性對工業發展產生不利影響,開發具有穩定孔結構、高比表面積和獨特表面化學性質的多孔吸附材料顯得尤為重要。對比碳基吸附材料、多孔金屬氧化物、沸石分子篩和MOF四種類型的吸附材料可以得出:
(1)碳基吸附材料來源廣泛、價格低廉,并且吸附范圍廣,但吸附過程多為不可逆,吸附劑的再生性有所欠缺;
(2)金屬氧化物相比于碳基吸附材料能夠連續使用且再生性好,但分離溫度相對較高;
(3)沸石分子篩孔道規整、選擇性較好,但吸附劑的高吸附能力和良好的再生性往往不能兼得;
(4)MOF 材料比表面積大、易于修飾、具有多種多樣的組成結構,相比于其他常規吸附劑,其吸附效率更高,但大多數MOF 的穩定性有待加強,在吸附過程中會出現孔道塌陷,材料設計制備與產業化研究仍在進行中。
對于百萬分之一含量H2S 的脫除,關鍵在于解決吸附劑內部傳質速率低和吸附活性不足的問題,同時還有吸附劑的吸附動力學。在不影響孔道結構和吸附容量的基礎上提高吸附動力學是目前深度脫硫的關鍵,基于此,具有特殊結晶結構金屬氧化物表現出極大的潛力。
干法深度脫硫在近幾年得到了廣泛的研究,并取得一定的研究進展。在實際應用中,需根據不同應用場景選擇不同的吸附劑。對于生產低硫油氣產品,天然氣、沼氣脫硫等石化領域,可以選擇碳基吸附劑以控制成本;對于生產高純度特種氣體、燃料電池原料氣等超深度脫硫領域,金屬氧化物、沸石分子篩及復合吸附材料則為更好的選擇。總而言之,精準理性設計并制備具有穩定孔結構、表面性質優良、吸附活性強、可再生利用的廉價吸附劑是深度脫硫的關鍵,也是今后研究的主要方向。隨著理論研究的不斷深入與合成方法的技術發展,相信高效價廉的深度脫硫吸附材料將會發揮更大的作用,推動石化行業的發展。
猜你喜歡
云南化工(2021年10期)2021-12-21 07:33:24
煤氣與熱力(2021年9期)2021-11-06 05:22:56
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
石油化工(2015年9期)2015-08-15 00:43:05
