新型磷酸二酯酶5抑制劑CPD1對肺動脈高壓大鼠血管收縮的影響
2021-03-11 06:51:00朱惠丹柯嘉琪王家璐徐甲甲劉曉晴楊建欽趙子建穆云萍李芳紅
中國藥理學通報 2021年3期
關鍵詞:效應
朱惠丹,柯嘉琪,王家璐,徐甲甲,劉曉晴,楊建欽,趙子建,穆云萍,李芳紅
(廣東工業大學生物醫藥學院,廣東 廣州 510006)
肺動脈高壓(pulmonary arterial hypertension,PAH)是一種進行性和致命性疾病,持續的血管收縮和血管重塑使得肺血管阻力(pulmonary vascular resistance,PVR)進行性增加,最終引起右心功能不全和衰竭,并導致死亡[1]。研究表明,肺動脈平滑肌細胞(pulmonary arterial smooth muscle cells,PASMCs)的收縮和增殖是肺血管收縮和重塑的主要原因,胞質內游離Ca2+濃度([Ca2+]i)的增加是PAH致病的關鍵因素[2],而胞內鈣穩態的維持與PASMCs膜上多種鈣通道的功能密切相關[3]。
磷酸二酯酶抑制劑是一種抑制磷酸二酯酶(phosphodiesterase,PDEs)活性的藥物,在心衰、哮喘、勃起功能障礙等疾病中具有廣泛的應用前景。研究表明,PDE5抑制劑[4]通過抑制PASMCs膜上鈣通道功能對肺血管重構和擴張有治療作用,顯著改善PAH患者的臨床狀態、運動能力和血流動力學參數[5-6]。目前,被批準用于治療PAH的PDE5抑制劑主要有他達拉非(Tadalafil)、西地那非等[7],但其表現出的頭痛、潮熱、視覺模糊等副作用仍未得到徹底解決,為了減少或者克服這些副作用,中國專利申請CN102020645A公開了一種吡唑并嘧啶酮衍生物(WYQ)的制備方法,提供了所述吡唑并嘧啶酮衍生物或其藥用鹽用于制備治療陽痿疾病及PAH藥物的用途,該藥物的特色是PDE5(IC50=0.001 4±0.000 1 μmol·L-1)的活性遠遠優于其他PDE同工酶,可有效降低頭痛等副作用[8]。盡管如此,該藥物水溶性差(室溫下低于2 g·L-1)的缺點不容忽視,基于此,團隊對WYQ開展了成鹽及共晶篩選,通過對發現的鹽型或共晶晶型樣品的理化性質評估,選出了水溶性有明顯提升(室溫下不低于13 g·L-1),且理化性質較優的晶型CPD1(中國專利申請號:201910505948.5,Fig 1),以滿足臨床用藥對于活性物質的形態、水溶性及純度等理化性質的嚴格要求。初步研究結果顯示,CPD1對PAH大鼠具有防治作用,但具體的作用機制還有待進一步研究。
因此,本研究使用野百合堿(monocrotaline,MCT)誘導的PAH大鼠模型,觀察CPD1對激動劑誘導的血管收縮效應的影響,闡明CPD1對平滑肌細胞中鈣通道功能的作用。為進一步探討CPD1對于PAH的作用機制奠定基礎。
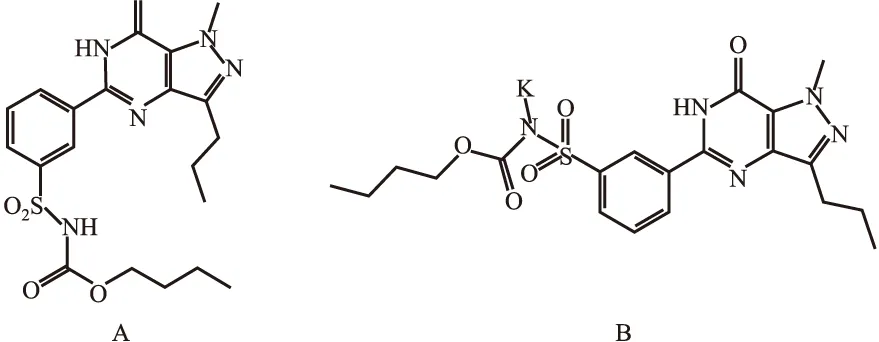
Fig 1 The structural formula of WYQ(A) and CPD1(B)
1 材料與方法
1.1 材料
1.1.1試劑 野百合堿(MCT,C2401)、氯化鉀(KCl,P5405)、氯化鎂(M2670)、氯化鈣(C1016)、HEPES(P005678)、苯腎上腺素(PHR1017)、內皮素-1(E7764)、乙酰膽堿(A6625)、二甲基亞砜(D2650),均購自美國Sigma公司,蘇木素伊紅染色試劑盒(C0105)購自上海碧云天生物技術有限公司。Tadalafil(171596-29-5)、CPD1(ET32637-37-P1)購自和委托藥明康德新藥開發有限公司合成,經COA分析測定CPD1純度為99%。MCT配制: HCl(1 mol·L-1)溶解固體藥物,充分溶解后用NaOH(3 mol·L-1)調節溶液pH到7.0,雙蒸水定容為2%溶液,現配現用;治療藥物配制:CPD1:用生理鹽水配成2.5 g·L-1的溶液;Tadalafil:用0.5%的羧甲基纖維素鈉配成2.5 g·L-1的溶液,均4 ℃冰箱保存。其他試劑均為國產分析純。
1.1.2儀器 多道生理信號采集處理系統(RM6240BDJ)、壓力換能器(YPJ01)、張力換能器(JZJ01H)、四腔器官浴槽系統(SQG-4)、離體腸管及熱板實驗恒溫裝置(HSS-1B)均購自成都儀器廠;解剖顯微鏡(SMZ645,日本Nikon);分析天平(BP 221S,德國Sartorius);酸度離子計(H1213A,意大利HANNA);石蠟包埋機(MPS/P1,德國SLEE);石蠟切片機(美國Thermo);冷凍臺(中威電子儀器有限公司);烘箱(DH6-9143B5-Ⅲ,上海新苗)。
1.2 方法
1.2.1MCT致PAH模型的建立 MCT作為一種誘導PAH模型的藥物在動物實驗研究中被廣泛使用[9],SPF級SD大鼠,♂,體質量(150±10)g,隨機分為正常對照組(12只)、MCT組(15只)、CPD1治療組(10只)、Tadalafil治療組(10只)。MCT組和治療組大鼠,以50 mg·kg-1的劑量給予一次性腹腔注射MCT,正常對照組則采用相同方法注射生理鹽水,常規飼養7 d后,治療組分別給予CPD1和Tadalafil灌胃,給藥量10 mg·kg-1·d-1,持續14 d,正常飲食飲水。
1.2.2右心室內壓測定 右心室壓力不僅能夠反映肺血管阻力的變化,而且對PAH預后有重要的提示作用,在利用動物模型研究PAH過程中,準確測量右心室壓力是檢測模型制備成功的關鍵技術[10]。啟動RM6240生理信號采集處理系統,選擇血壓測量項,調節血壓單位、坐標軸位置及走紙速率等參數,穩定30 min后用水銀柱血壓計定標壓力換能器。大鼠用烏拉坦(0.5 g·kg-1)腹腔麻醉并進行腹腔肝素化(500 IU·kg-1)處理,迅速經右頸外靜脈行右心室(right ventricle,RV)插管術,記錄各組大鼠的右心室收縮壓(right ventricular systolic pressure,RVSP),壓力曲線穩定后持續記錄5-10 min后結束測量實驗。
1.2.3右心室質量指數測定 RVSP測定結束后嚴格按照華南理工大學實驗動物中心的規定對大鼠實施安樂死,迅速取出胸主動脈、心、肺組織,分離右心室(right ventricle,RV),濾紙吸干血液和水分,分析天平稱重。同法稱取左心室(left ventricle, LV)與室間隔(interventricular septum,VS)重量,計算各組大鼠的右心室質量指數(right ventricular mass index,RVMI)[3]:RVMI=RV/(LV+VS)。
1.2.4肺動脈形態學檢測 大鼠安樂死后,連同氣管將心、肺取出,左肺經中性甲醛(10%)固定24 h、脫水后,按常規石蠟包埋切片步驟,在最大縱切面連續切片,取厚度為4-5 μm肺組織4片,行蘇木精-伊紅染色。在LEICA光學顯微鏡下觀察各組大鼠肺小動脈(直徑:50-150 μm)形態,每張肺切片至少選取5個視野尋找肺小動脈拍照,對管壁厚度進行測量,計算管壁厚度和厚度百分比(WT%)。
1.2.5血管張力檢測 應用離體血管張力檢測技術觀察血管張力的變化[3]。體視顯微鏡下分離出胸主動脈和肺內動脈,并將其剪成約3-5 mm的血管環,三角鉤滾動去內皮后動脈環置于四腔浴槽中,給以1 g負荷,經張力換能器轉換后在RM6240BDJ多道生理信號記錄儀上描記張力曲線,調節基線,平衡2 h。實驗前先給予KCl 60 mmol·L-1收縮血管,重復3次,判斷血管活性,當前后連續2次得到的收縮幅度差小于10%,被認為標本反應可重復,否則棄之。血管環加入苯腎上腺素(phenylephrine,Phen,終濃度0.1 μmol·L-1),待預收縮反應達平臺穩定后,加入乙酰膽堿(acetylcholine,Ach,10 μmol·L-1),觀察血管環的舒張效應,驗證去內皮程度,血管舒張明顯超過Phen收縮效應20%的棄之,本研究中血管環活性和去內皮合格率保持在80%-90%。隨后,觀察內皮素-1(endothelin-1,ET-1)在各組大鼠肺動脈和主動脈中的收縮效應,為避免血管長度和直徑的差距對統計結果產生不可避免的影響,各種藥物誘發血管的收縮作用以第3次60 mmol·L-1KCl收縮效應的百分數來標準化。
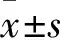
2 結果
2.1 CPD1對MCT致PAH大鼠模型形成的影響
2.1.1對RVSP的影響 與正常對照組(CON)相比,MCT組大鼠的RVSP升高,表明MCT可致大鼠右心室內壓升高,肺動脈阻力增高,PAH模型構建成功。CPD1治療后可抑制模型鼠RVSP的升高,且抑制效果不低于陽性對照藥Tadalafil,表明CPD1能夠降低MCT引起的高右心室壓力,見Fig 2 A-E。
2.1.2對RVMI的影響 與CON相比,MCT組大鼠的RVMI升高,表明MCT可誘導大鼠右心室肥厚。CPD1干預后,大鼠的RVMI降低,同樣抑制效果不低于Tadalafil,表明CPD1能減輕MCT誘導的大鼠右心室肥厚,而各組大鼠的心率沒有差別(Fig 2F,G)。結果提示CPD1干預能有效抑制MCT誘導的大鼠PAH和右心室肥厚。

Fig 2 Effect of phosphodiesterase type 5 inhibitors on RVSP and RVMI in MCT rats
2.1.3對肺小動脈形態學的影響 與CON相比,MCT組大鼠肺內小動脈的中膜厚度和厚度百分比均明顯增大,表明MCT可致大鼠肺內動脈平滑肌細胞增生,肺血管重構。CPD1和Tadalafil干預后,大鼠肺內小動脈中膜變薄,中層平滑肌增生減少,管腔增大,見Fig 3。結果提示: CPD1干預能有效抑制MCT誘導的大鼠肺動脈增殖和重塑。
2.2 CPD1對PAH大鼠肺動脈和主動脈收縮效應的影響
2.2.1血管環去內皮程度檢測 Phen作用后血管明顯收縮,觀察Ach的舒血管效應,結果顯示:未破壞內皮的肺動脈血管環中,Ach誘導血管顯著舒張,表明血管內皮完整;而去內皮后Ach的舒血管效應明顯減弱,兩者比較差異有顯著性(P<0.01),表明去內皮成功,標本可用,實驗者去內皮手法可靠,見Fig 4。
2.2.2對Phen引起的血管收縮效應的影響 觀察CPD1干預后對Phen(0.1 μmol·L-1)引起的主動脈和肺動脈收縮效應的影響。結果顯示:與CON相比,MCT大鼠中Phen引起的肺動脈收縮效應顯著增強,CPD1治療組的收縮效應減小,見Fig 5A-D;此外,相比MCT組,MCT+CPD1組Phen引起的主動脈收縮效應也減小,見Fig 5E-H。提示CPD1干預能抑制PAH大鼠肺動脈對Phen的高反應性,CPD1可能通過調控血管平滑肌細胞內鈣通道功能發揮作用。
2.2.3對KCl引起的血管收縮效應的影響 為進一步評價鈣通道在CPD1治療PAH的作用,繼續觀察各組大鼠血管對60 mmol·L-1KCl的收縮效應。結果顯示:與CON相比,MCT組大鼠中KCl引起的主動脈和肺動脈收縮效應均無變化,表明MCT致PAH對平滑肌細胞電壓依賴性鈣通道(voltage-dependent calcium channel,VDCC)功能無明顯影響,見Fig 6。此外,CPD1治療后KCl引起的主動脈和肺動脈收縮效應也無明顯變化,表明CPD1干預對PAH大鼠平滑肌細胞中VDCC作用不明顯。
2.2.4對ET-1誘導的血管收縮效應的影響 繼續觀察各組大鼠血管對ET-1(10 nmol·L-1)的收縮效應。ET-1是一種強血管收縮劑,可激活非電壓依賴性鈣通道,主要是鈣池操縱性鈣通道(store operated Ca2+channel,SOCC)和受體操縱性鈣通道(receptor-operated Ca2+channels,ROCC),使血管收縮和增殖[11]。結果顯示:相比CON組,MCT組的ET-1收縮肺動脈效應增高,見Fig 7A-D。提示PAH大鼠肺動脈對ET-1的高反應性;相比MCT組,CPD1治療組肺動脈和主動脈的收縮效應均減小,見Fig 7D,H。表明CPD1干預能抑制PAH大鼠肺動脈和主動脈的非電壓依賴性鈣通道活性,且可能是CPD1抑制肺動脈ET-1高反應性作用的重要因素。
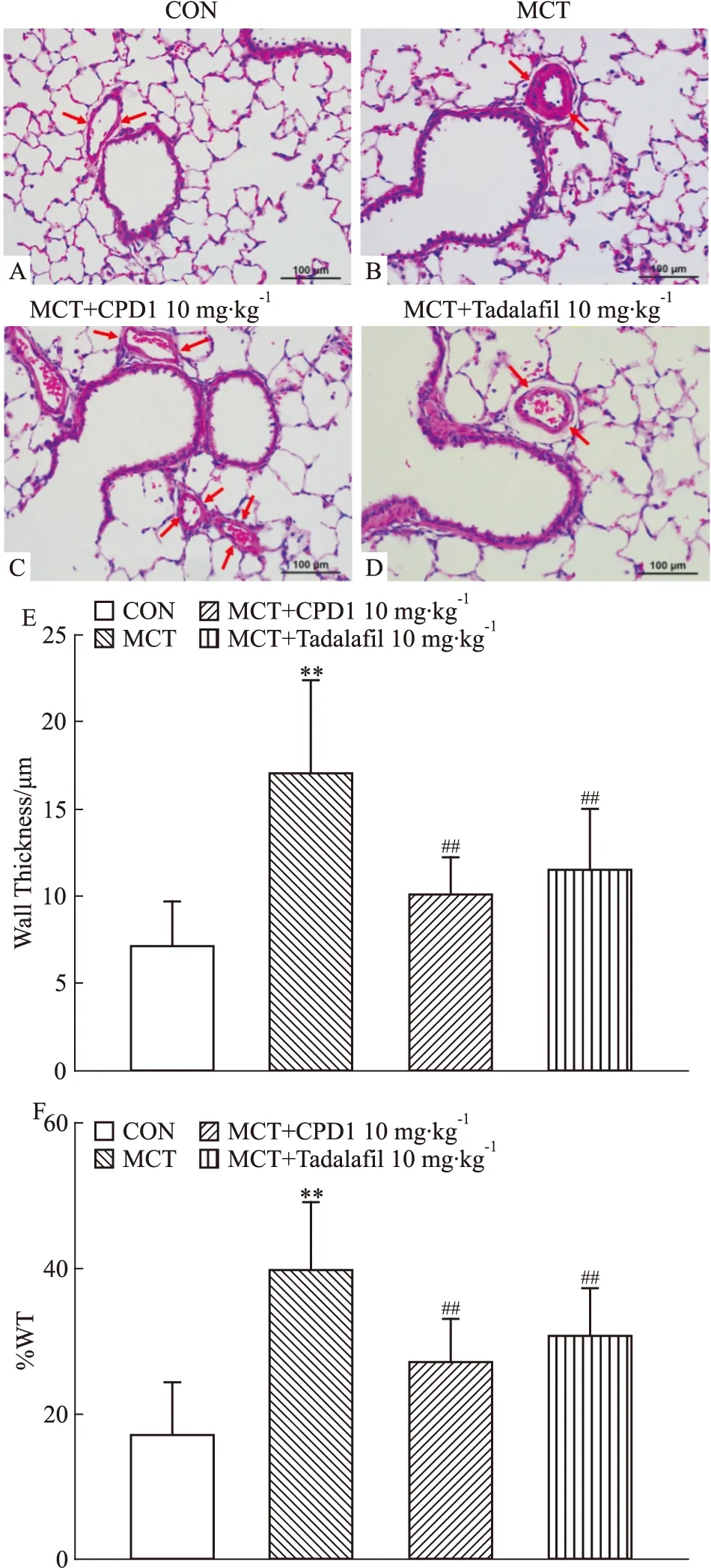
Fig 3 The representative images ofhematoxylin-and eosin stained lung section fromcontrol,MCT, CPD1 and Tadalafil-treated rats,respectively,showing small pulmonary arteries(radius:101-150 μm)control: A section(×200), MCT: B section(×200), MCT+CPD1: C section(×200); MCT+Tadalafil: D section(×200); and the ratio of vessel wall thickness(E) and the percentage of vascular wall thickness/ vascular external diameter(WT%)(F) in PAs of control(n=36), MCT(n=30), CPD1-pretreated(n=31) and Tadalafil-pretreated(n=31) rats.**P<0.01 vs control;##P<0.01 vs MCT.
Fig 4 Ach induced relaxation of pulmonary arteries(PAs) precontracted by phenylephrine(Phen)
2.3 CPD1對正常大鼠肺動脈和主動脈的作用為了明確CPD1對大鼠血管作用的普遍性和機制,繼續觀察CPD1預處理對ET-1(10 nmol·L-1)收縮正常大鼠血管的作用。結果顯示:CPD1預處理可抑制ET-1對正常大鼠肺動脈和主動脈的收縮效應,見Fig 8。提示CPD1對正常大鼠血管收縮效應的舒張作用可能主要是通過阻斷非電壓依賴性鈣內流產生的。
3 討論
心血管疾病依然是全球死亡主要原因[12],PAH作為一類危及生命的肺部疾病,有著心血管疾病的“惡性腫瘤”之稱,PVR的持續性增加導致右心功能不全、右心衰竭乃至死亡。目前,已批準用于治療PAH的靶向藥物主要包括前列環素類、內皮素受體拮抗劑和PDE5抑制劑[13]。PDE5抑制劑能夠抑制PDE5,增加細胞內cGMP水平,激活蛋白激酶G(PKG),誘導內源性NO的舒血管作用,達到降低肺動脈高壓的目的。美國食品藥品管理局已批準西地那非和Tadalafil用于治療PAH,研究表明,西地那非可改善高原運動時的血流動力學和運動性能參數[7],但因其半衰期短,需每天多次大劑量用藥(100 mg·d-1),加重了患者的經濟和身體負擔;Tadalafil 10 mg(2次·d-1)可降低高原缺氧性PAH肺水腫的發生率,但Tadalafil幾乎不溶于水,可能影響其生物利用度。靶向治療藥物出現之前,特發性PAH患者中位生存期僅為2.8年,近年來,盡管治療方案有了顯著進展,但仍不能從根本上“治愈”PAH,其3年生存率只有68%[14],且治療藥物價格昂貴,副作用明顯。為此,亟需尋找可靠的治療藥物以減輕患者負擔,提高PAH的總體生存率。
為了克服現有PDE5抑制劑的缺點,中國專利文獻(CN102020645A)公開了一種吡唑并嘧啶酮衍生物WYQ,具有與西地那非和Tadalafil相同的療效,且具有低劑量高藥效的特點[8]。基于此,團隊自主研發獲得一種新型PDE5抑制劑CPD1,與WYQ結構相似(Fig 1),但水溶性和腸溶性更好、毒性低、理化性質更穩定。本研究利用MCT致PAH大鼠模型,觀察了CPD1對PAH大鼠的治療作用,以及對不同鈣通道激動劑誘發的血管收縮效應的影響。結果顯示CPD1干預后,降低PAH大鼠的RVSP和RVMI,且使肺內小動脈管壁內膜變薄,管腔增大,說明CPD1干預能減輕PAH大鼠右心室壓力增高和右心室肥厚,逆轉肺內小動脈的增殖和重塑,提示CPD1干預能抑制MCT致PAH大鼠模型,此外,CPD1干預后,能夠降低PAH引起的大鼠死亡率,但如果延長給藥時間,藥物作用是否依然存在持久性,需要進一步的實驗研究。且與Tadalafil治療組相比,CPD1抑制PAH可能有更好的效果。但CPD1干預對PAH大鼠血管平滑肌細胞中鈣通道功能的作用還不明確。
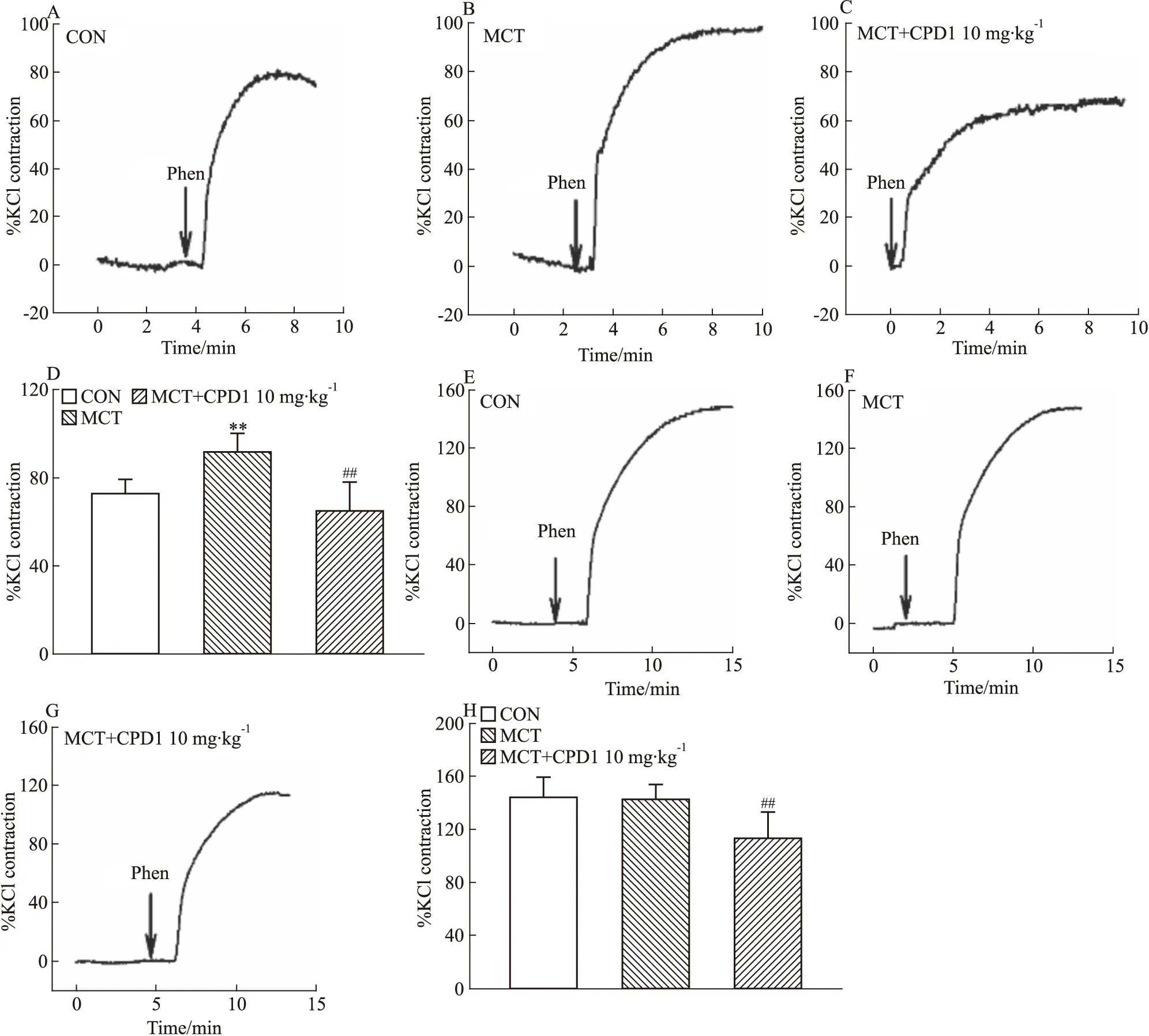
Fig 5 Phen-induced ring contraction in MCT-induced and CPD1-pretreated MCT-induced rats The representative traces of Phen-induced PAs ring contraction in CON(A), MCT-induced(B) and CPD1-pretreated MCT-induced(C) rats. The representative traces of Phen-induced aorta ring contraction in CON(E), MCT-induced(F) and CPD1-pretreated MCT-induced(G) rats. The average values of Phen-induced PAs ring(D) and aorta ring(H) contraction in CON(n=13 and 11), MCT-induced(n=11 and 8) and CPD1-pretreated MCT-induced(n=10 and 10) rats.**P<0.01 vs control,##P<0.01 vs MCT.
PASMCs膜上主要存在3種鈣通道:VDCC、SOCC和ROCC,三者共同參與維持胞內鈣穩態的平衡。肺血管的收縮主要與PASMCs收縮相關,而PASMCs的收縮主要依賴于膜上鈣通道介導的鈣內流,使[Ca2+]i升高。研究已證實,cGMP可負向調節鈣信號傳導和鈣通道活性[15],西地那非通過上調VDCC抑制低氧誘導的人PASMCs增殖[16],為進一步明確CPD1對PAH大鼠血管平滑肌細胞中不同鈣通道功能的作用,在CPD1干預MCT致PAH大鼠模型的基礎上,觀察CPD1對不同激動劑誘發的血管收縮效應的影響。本研究結果發現,CPD1干預可下調Phen引起的肺動脈和主動脈收縮,但對VDCC特異性激動劑KCl引起的收縮效應無影響,表明CPD1干預對血管平滑肌細胞中VDCC功能影響不明顯。ET-1由血管內皮細胞產生,主要通過激活SOCC和ROCC,引起血管收縮[11],在PAH患者ET-1受體表達增加,且ET-1濃度增加,與肺血流量和心輸出量成反比關系。本研究發現,CPD1干預能抑制MCT致PAH大鼠肺動脈對ET-1的高反應性,顯著減小主動脈上ET-1的收縮效應,提示CPD1干預能抑制PAH大鼠肺動脈和主動脈中非電壓依賴性鈣通道的功能,且可能是CPD1發揮抑制MCT致PAH作用的重要因素。盡管多項研究表明,PDE5抑制劑對血壓影響不明顯[17],但本研究發現CPD1預處理血管環,抑制正常大鼠中ET-1誘發的主動脈收縮效應,表明PDE5抑制劑可能對主動脈平滑肌細胞膜上的非電壓依賴性鈣通道功能同樣有抑制作用,這對于PDE5抑制劑的臨床用藥具有指導意義。本工作闡明了新型PDE5抑制劑CPD1與血管平滑肌細胞膜上鈣通道功能之間的關系,對于針對PAH的新藥研究和臨床用藥具有重要意義。
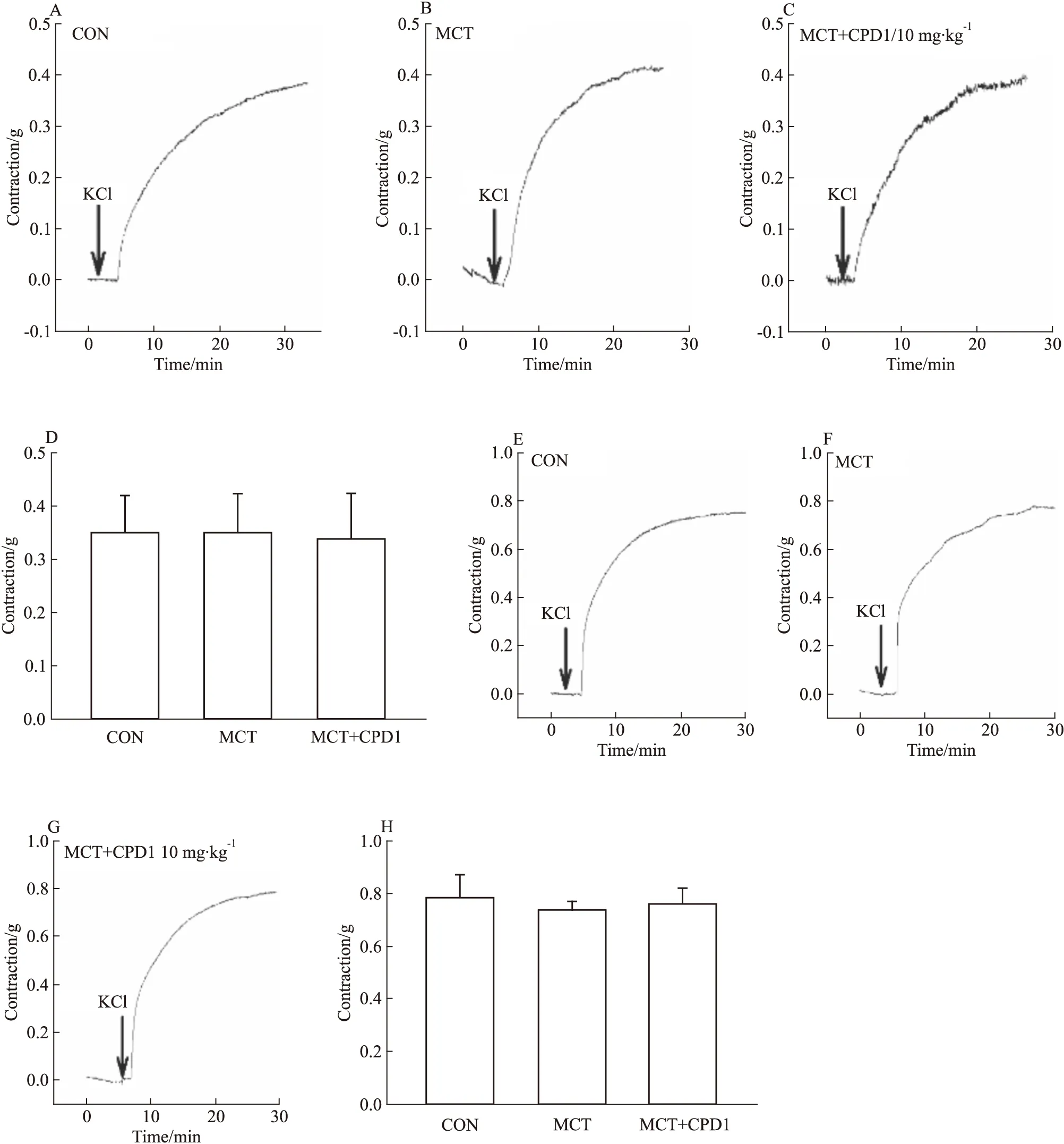
Fig 6 KCl-induced ring contraction in MCT-induced and CPD1-pretreated MCT-induced rats The representative traces of KCl-induced PAs ring contraction in CON(A), MCT-induced(B) and CPD1-pretreated MCT-induced(C) rats. The representative traces of KCl-induced aorta ring contraction in CON(E), MCT-induced(F) and CPD1-pretreated MCT-induced(G) rats. The average values of KCl-induced PAs ring(D) and aorta ring(H) contraction in CON(n=14 and 12), MCT-induced(n=13 and 8) and CPD1-pretreated MCT-induced(n=12 and 12) rats.
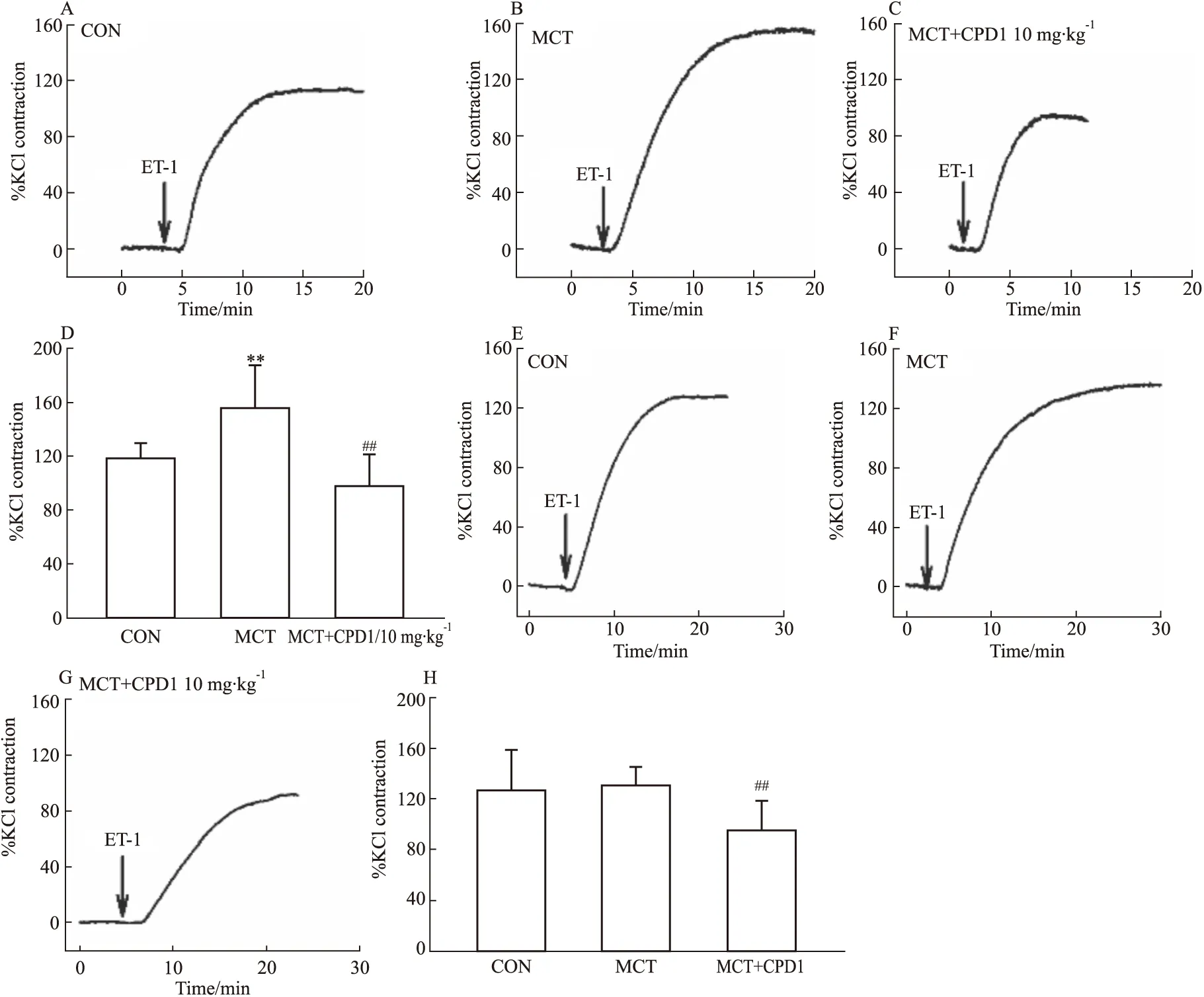
Fig 7 ET-1-induced ring contraction in MCT-induced and CPD1-pretreated MCT-induced rats The representative traces of ET-1-induced PAs ring contraction in CON(A), MCT-induced(B) and CPD1-pretreated MCT-induced(C) rats. The representative traces of ET-1-induced aorta ring contraction in CON(E), MCT-induced(F) and CPD1-pretreated MCT-induced(G) rats. The average values of ET-1-induced PAs ring(D) and aorta ring(H) contraction in CON(n=12 and 9), MCT-induced(n=14 and 14) and CPD1-pretreated MCT-induced(n=16 and 10) rats.**P<0.01 vs control,##P<0.01 vs MCT.
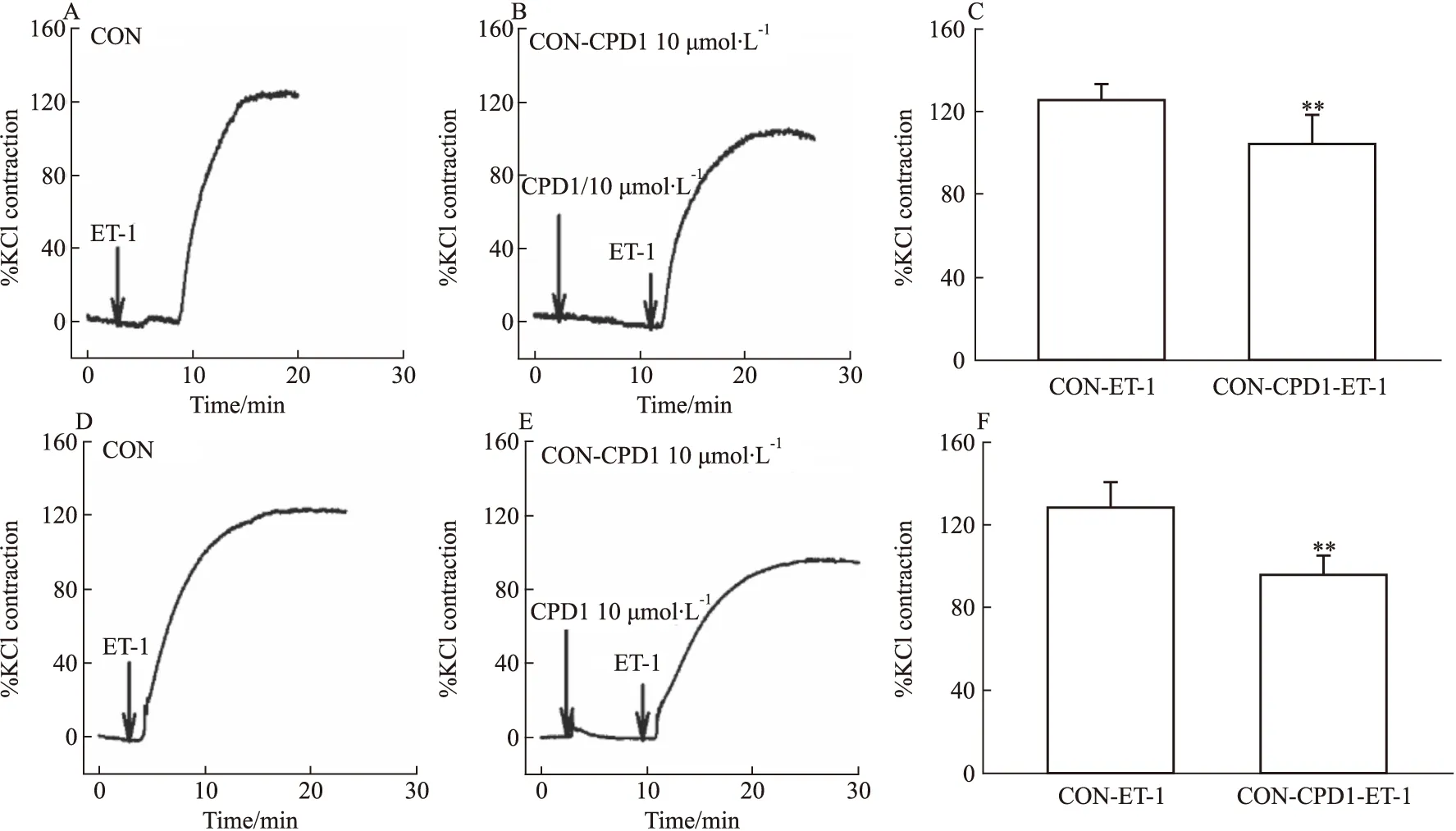
Fig 8 Effect of CPD1-pretreated on ET-1-induced ring contraction in control rats
猜你喜歡
核科學與工程(2021年4期)2022-01-12 06:30:26
今日農業(2020年19期)2020-12-14 14:16:52
小學生必讀(中年級版)(2020年9期)2020-12-04 02:07:22
科學大眾(2020年17期)2020-10-27 02:49:10
紅土地(2018年11期)2018-12-19 05:10:56
意林·全彩Color(2018年9期)2018-11-13 22:49:38
中學物理·高中(2016年12期)2017-04-22 11:53:03
中國衛生(2016年4期)2016-11-12 13:24:14
中國衛生(2014年4期)2014-12-06 05:57:14
小櫻桃·童年閱讀(2014年11期)2014-12-01 22:21:30
