甲烷活化與催化轉化過程中的幾個關鍵問題及對策
2021-03-21 04:53:46肖曉敏曹學雨吳新宇耿豪杰於思瑜劉社田
工業催化 2021年12期
關鍵詞:催化劑
肖曉敏,曹學雨,吳新宇,耿豪杰,於思瑜,劉社田
(西南大學化學化工學院,重慶 400715)
甲烷是最簡單的有機物,也是含氫量最大的烴,主要以天然氣、頁巖氣、煤層氣和甲烷水合物的形式存在。由于其在地殼中蘊藏量豐富,是短期內可以有效取代煤炭、石油的清潔能源和化工原料。甲烷作為化工原料可以生產有機材料、液體燃料及其它化學品。由于甲烷分子中氫含量遠高于大多數化學品,脫氫是甲烷轉化的關鍵步驟,甲烷轉化為化學品可以副產大量的氫氣。因此,甲烷也是氫能產業發展中值得特別重視的能源資源。通過提高甲烷的使用量,可以有效地降低二氧化碳的排放,有利于達成國家制定的碳達峰和碳平衡的能源戰略目標。本文主要分析了實現甲烷活化和高效直接轉化過程中的一些關鍵技術問題,主要包括臨氧轉化過程中的選擇性氧化、脫氫低聚過程中的熱力學平衡限制及催化劑穩定性等,并對其可能的解決途徑進行了探討。
1 甲烷活化與轉化過程
甲烷分子擁有特殊的正四面體結構,其C—H鍵能達到439.4 kJ·mol-1[1]。這使得甲烷在常規條件下難以活化,通常需要在強酸強堿或高溫條件下進行,且生成的產物活性或反應性能遠高于甲烷,易進一步轉化,導致目的產物選擇性難以控制。這是從甲烷合成高附加值化學品所面臨的核心挑戰。
通常甲烷分子的活化需要催化劑的作用,活化機理可以分為以下三類:(1)C—H鍵與催化劑形成金屬-碳σ鍵的中間體或最終產物;(2)C—H鍵僅與催化劑作用,金屬-碳σ鍵不在任何階段直接產生;(3)催化劑首先與另一種反應物形成活性中間物,然后攻擊甲烷分子使C—H鍵活化。有關甲烷活化機理方面的論述可見文獻[2-3]。這里主要討論在工業上有重大應用價值和前景的甲烷轉化過程。一般來講,甲烷的轉化途徑可以分為間接轉化與直接轉化兩種。
甲烷的間接轉化過程是將甲烷首先轉化為不同CO/H2比例的合成氣(CO+H2),再進一步合成需求的產品。工業上利用甲烷合成化學品主要通過間接轉化來實現,包括大規模制氫、合成氨[4]、合成醇[5]和費托合成等[6]。甲烷通過蒸汽重整(SRM,式1)制備合成氣是成熟的工業技術,但仍存在催化劑結焦、中毒失活等問題[7-8],所以依然是催化領域的重要研究課題。甲烷蒸汽重整是強吸熱過程,也是眾多甲烷間接轉化過程中能耗最大的步驟。因此,為了進一步提高過程能效,研究者也在積極開發部分氧化(POM,式2)、干氣重整(DRM,式3)、自熱重整(ATR)和聯合重整(CRM)等技術和工藝[9-10]。其中面臨的技術難題主要是催化劑的開發,以及催化劑抗積碳、抗活性金屬的燒結、抗中毒等性能的提高[11-12]。

(1)

(2)
(3)
目前,重整反應的催化劑主要是以貴金屬催化劑和摻雜貴金屬的鎳基催化劑為主。甲烷間接轉化過程的工藝流程比較復雜,生產設備投資較大,能源利用率較低,這是工業上希望開發直接轉化技術的主要原因。
甲烷直接轉化是通過一步反應直接制備目的產物的過程,主要包括甲烷氧化偶聯(OCM)、甲烷選擇性氧化(SOM)以及甲烷脫氫低聚(DOM),主要產物為烯烴、炔烴和芳烴等。已經工業化的直接轉化過程有甲烷高溫裂解生產乙炔[13]、炭黑,甲烷氨氧化生產氫氰酸[14]等。這些工業化的過程生產規模相對較小。而通過甲烷轉化直接生產乙烯、甲醇或芳烴等大宗化工原料,工業上急切期盼的低能耗過程卻都在實驗室技術開發階段,還需要大量的基礎研究工作。
甲烷氧化偶聯制乙烯技術(式4)于1982年由Keller G E等[15]首次提出,并在全球范圍內引發了研究者的廣泛研究。典型的、性能較為優良的催化劑包括Na2WO4/Mn/SiO2[16]和Li/MgO[17]。但催化劑的性能特別是單程反應C2(乙烯+乙烷)產物收率和催化劑穩定性仍無法滿足工業化要求。
(4)
甲烷在無氧條件下的轉化可以避免生成CO2和H2O等副產物,從而提高碳原子利用率。1993年,Wang L等[18]首次報道了在Mo/HZSM-5催化劑上甲烷有效轉化成苯的反應(式5),從而在全球引發了一波針對甲烷直接脫氫低聚制備芳烴催化劑的研究。HZSM-5、MCM-22分子篩負載Mo、Re等活性金屬組分的催化劑對該反應表現出良好的催化活性。
(5)
甲烷選擇性氧化制甲醇無論從清潔能源生產還是化學品合成考慮都是工業上夢寐以求的過程。有研究預測[19],利用氣固相催化技術,分子氧作氧化劑,甲烷單程轉化率達到5.5%,甲醇選擇性達到80%就可以和工業上經合成氣合成甲醇的過程進行競爭。然而,文獻報道的單程甲醇收率大多低于2%。所采用的催化劑活性組分多為V、Mo、Fe等的氧化物。
2CH4+O2→ 2CH3OH+ΔH
(6)
2 活化與催化轉化過程中關鍵問題及對策
經過幾十年的研究與發展,甲烷活化與催化轉化的相關基礎和應用研究已十分深入,但仍存在一些關鍵技術難題,致使甲烷轉化效率較低,無法達到工業化過程的要求。這些技術難題包括臨氧轉化過程中的選擇性氧化問題、脫氫低聚過程中的熱力學平衡限制、高溫反應條件下催化劑失活和材料的穩定性問題。
2.1 臨氧轉化過程中的選擇性氧化
甲烷臨氧轉化過程包括甲烷催化重整制合成氣、甲烷氧化偶聯制乙烯、甲烷選擇性氧化制甲醇等。其中甲烷重整制合成氣,包括蒸汽重整、CO2干重整、部分氧化自熱重整等是比較成熟的工業化技術。基于熱力學原理考慮,該技術實現工業化的主要原因之一是產品合成氣CO+H2在反應體系中是相對穩定的目的產物,并和反應物CH4+H2O處在接近化學平衡的狀態,且轉化過程是熱力學有利的。反觀甲烷氧化偶聯制乙烯和甲烷選擇性氧化制甲醇,盡管反應在熱力學上有利,但在大多數研究的反應條件下[前者(700~900) ℃,后者(300~600)℃]反應的目的產物乙烯或甲醇不穩定,極易和反應物中的O2發生深度氧化反應生成熱力學上更加穩定的CO2或CO。這一理論事實決定了如何在反應過程中保持目的生成物乙烯或甲醇的相對穩定,并使其及時脫離反應體系成為獲得較高目的產物產率的關鍵。
多數文獻認為,甲烷在氧化偶聯催化劑上的活化可以分為兩類:一類是分子氧在催化劑表面首先被活化(如在Li/MgO上)產生親電活性氧物種如O-或O2-,甲烷分子與這些親電氧物種作用發生C—H鍵均裂生成CH3·,CH3·在氣相發生偶聯生成C2H6進而脫氫生成C2H4;另一類是甲烷分子吸附在路易斯酸-堿對上(如在堿性La2O3或MgO表面)發生C—H鍵激化異裂生成甲基陽離子或在分子氧作用下生成CH3。研究表明,甲烷在路易斯酸-堿中心上的活化與中心的酸堿強度和局部結構有關。甲烷氧化偶聯實質是甲烷脫氫偶聯生成C2H6/C2H4的吸熱反應與氫氣氧化放熱反應的耦合過程。這種反應的耦合使得甲烷脫氫生成乙烯的過程由熱力學不利轉化為熱力學有利。然而,由于產物或中間活性碳物種的深度氧化反應,無法同時獲得較高的甲烷轉化率和產物選擇性,使產物收率受到限制。目前,甲烷氧化偶聯反應的C2收率及選擇性仍無法達到工業化要求。如要獲得較高的乙烯收率,一方面需要催化劑上表面活化反應保持高度的選擇性,避免碳氫表面物種的深度脫氫和氧化;另一方面也需要避免中間產物在氣相或催化劑表面的深度氧化反應。
甲烷選擇性氧化制甲醇是另一個放熱且熱力學有利的反應,也是工業上夢想的大規模直接液化甲烷的催化過程。早期研究最多的是Mo-、V-氧化物催化劑[20-21]。此類催化劑上甲烷通過與催化劑表面的MOx相互作用形成H-M(Ox)-CH3進而轉化為甲醇。而近期許多研究則主要集中于模仿甲烷單加氧酶的分子篩負載Fe、Cu催化劑體系[22- 23]。為獲得較高的產物收率常使用H2O2代替分子氧,如Cui X等[24]報道的使用石墨烯限域的鐵單原子催化劑上CH4低溫選擇性氧化反應。該類催化劑與Mo-、V-氧化物類似,即首先在金屬中心形成活性氧中間物種,進而與甲烷分子作用使甲烷C—H鍵活化。目前固體催化劑上甲烷選擇性氧化制甲醇所面臨的最大挑戰是無法同時獲得較高的轉化率和產物選擇性。Latimer A A等[25]最近提出一個動力學模型,認為甲烷選擇氧化制甲醇連續反應過程中甲醇收率受本征動力學限制。因此,開發溫和反應條件下的氧化過程,并對生成的甲醇予以“保護”是未來的研究需要重視的[26]。
2.2 脫氫低聚過程中的熱力學平衡限制
甲烷脫氫是強吸熱反應,在不同溫度下甲烷轉化為乙烯和苯的平衡轉化率如圖1所示。由于苯分子的特殊電子結構和對稱性,使得其在高溫條件下比乙烯更為穩定,在一定反應溫度下甲烷轉化為苯可獲得更高的轉化率。但即使在973K的反應溫度下,甲烷的平衡轉化率也只有11.4%,屬于熱力學上不利的反應。事實上,文獻研究所獲得的甲烷轉化率只有8%~10%,與熱力學計算結果吻合。在圖1中也給出了甲烷裂解完全脫氫反應的平衡轉化率。隨著體系反應溫度的升高,甲烷向積碳轉化的平衡轉化率遠遠高于生成乙烯或苯的平衡轉化率,這使得反應體系中的積碳反應難于避免,成為阻礙甲烷高效轉化為烴類產物的主要難題。值得指出,即使在高溫反應條件下,單程反應也無法獲得較高的甲烷轉化率,甲烷直接芳構化過程在工業上的應用必然面臨產物和反應物分離所導致的高能耗和高生產成本的問題。
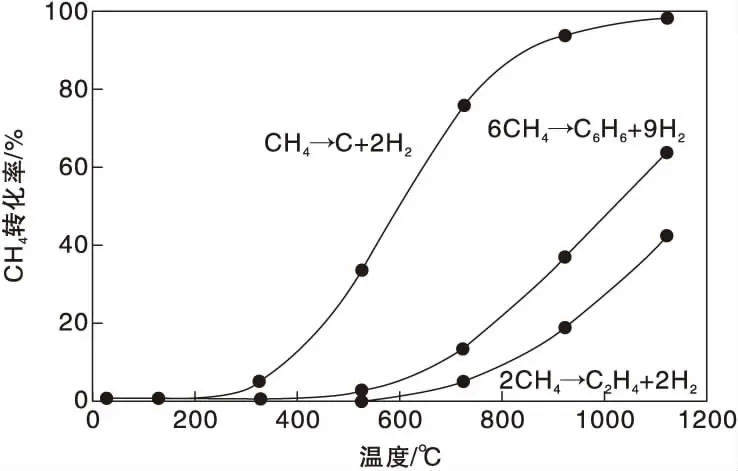
圖1 無氧條件下甲烷催化轉化反應的甲烷轉化率[27]
2.3 高溫反應條件下脫氫低聚催化劑的穩定性
在甲烷脫氫低聚反應中,隨著反應時間和反應溫度的增加,烴類熱裂解易產生大量積碳,積碳主要覆蓋在催化劑的活性位上和分子篩孔道中,導致其快速失活,反應活性降低。這是有關甲烷脫氫低聚生產烯烴或芳烴所面臨的另一方面技術難題。研究表明,在Mo/HZSM-5催化劑上甲烷脫氫芳構化反應主要生成三種類型的表面積碳:(1)主要分布于分子篩孔道中的一種不定型或石墨型的積碳;(2)主要分布于分子篩外表面的Mo2C中的碳元素;(3)貧氫的sp型(稠環芳烴)積碳。在甲烷活化過程中,貧氫型積碳會隨著反應時間的推移而在催化劑表面逐漸累積,直至完全覆蓋活性金屬和分子篩表面。針對積碳問題,研究者使用脫鋁、表面硅烷化、構建介孔結構等多種手段對催化劑進行改性研究。Ding W等[28]使用大量的外來硅源選擇性地覆蓋和消除沸石的外表面酸性位點,可有效降低HZSM-5外表面酸性位點的密度,提高單環芳烴產率,降低催化劑失活速率。Liu H等[29]通過實驗證實,HZSM-5分子篩表面經硅烷化處理后,能夠減少表面沉積的碳物種量,從而使得催化劑反應活性提高。通過創建介孔,Wu Y等[30]將Mo浸漬到多層載體材料MFI中合成中孔/微孔沸石催化劑。結果表明,中孔結構使反應物向孔道內活性位點的擴散更易發生,因此在Mo/MFI催化劑上可獲得更高的甲烷轉化率和芳烴產率。但催化劑改性只能改善甲烷轉化率和積碳問題,無法從根本上根除積碳反應并使產物的生成突破反應熱力學平衡的限制。
2.4 提高烴類產品的策略
甲烷脫氫低聚生產烯烴或芳烴過程面臨高溫反應條件下產物平衡產率低以及催化劑在高溫反應條件下積碳失活等技術難題。因此,如何在較溫和條件下實現較高單程反應轉化率并保持催化劑一定的穩定性成為未來研究開發需要思考的問題。解決這一矛盾的對策包括:采用膜反應器將脫氫反應與氫分離過程耦合提高甲烷轉化率,以及利用氧化劑或氫捕獲劑降低反應體系氫分壓從而提高目的產物收率。采用膜分離反應器的有效性已被許多研究工作所證實[31-32]。但甲烷轉化率的提高卻受到所采用分離膜材料氫分離性能的限制。最近,Kumar A等[33]報道在甲烷芳構化過程中添加氫吸收劑(Zr)使得甲烷在973 K下的轉化率由10%提高到27%。毫無疑問,這種分離氫的方式比采用氫分離膜更加有效,為脫氫過程的研究提供了新思路。又例如,Kikuchi E等[34]將Pd或Pd-Ag復合膜鍍在多孔氧化鋁陶瓷的外表面,突破了甲烷重整制氫的熱力學平衡,實現了氫氣選擇性增強的效果。研究證明在500 ℃、101.325 kPa條件下Pd膜及Pd-Ag復合膜的甲烷轉化率能達到60%以上,而理論平衡值僅為42%,并且甲烷轉化率隨Pd膜厚度降低而提高。Cao Z等[35]利用鈣鈦礦Ba0.5Sr0.5Co0.8Fe0.2O3-δ(BSCF)膜研究甲烷芳構化反應,發現通過氧滲透膜滲入的氧氣不會破壞催化劑活性組分Mo2C。在長達1000 min的反應后,與固定床反應器相比,膜反應器中的甲烷轉化率高出3%,芳香族化合物選擇性高出30%,并且生成的水蒸氣能有效抑制積碳,積碳量僅占廢催化劑的1.2%。Morejudo S等[36]設計了BaZr0.7Ce0.2Y0.1O3-δ(BZCY72)膜,使氧離子迅速向反應介質傳輸將H2氧化為H2O,同時操作過程中產生了少量的CO,兩者都降低了催化劑表面的結焦行為并提高了芳烴選擇性。
3 結 語
實現甲烷在固體催化劑作用下的高效定向轉化的前提條件是催化劑上的活性中心應具備適當的化學環境和空間環境,以有利于C—H鍵的斷裂,或者能夠產生活性中間物種進而與甲烷分子作用導致C—H鍵斷裂。然而僅僅通過對C—H鍵的活化還難以達到甲烷分子定向轉化為C2+烴類或甲醇等含氧有機分子的目的。碳氫鍵活化后所產生的CH3、或CH2等活性中間物種必須發生相互作用生成C—C鍵或與第二種反應物(如分子氧或其活化后的中間物種)發生相互作用生成C—O鍵才可能生成目的產物。這種活性中間物種之間及其與催化劑活性中心之間的相互作用是決定甲烷優勢轉化方向的關鍵步驟。因此可以推測,在甲烷脫氫低聚生成C2+烴類產品過程中,形成C—C鍵的優勢途徑取決于CHx活性中間物種的相對穩定性(減少深度脫氫)和其在局部空間或催化劑表面上的濃度。因此通過構筑適當的活性中心實現C—H鍵的活化,調控CHx與活性中心的相互作用并保持其適當的穩定性才有利于C—C的形成,同時也有利于抑制深度脫氫積碳反應的發生。另一方面,在臨氧反應過程中,無論是甲烷氧化偶聯還是選擇性氧化制甲醇,都需要考慮如何避免目的產物深度氧化的問題。事實上,甲烷臨氧轉化過程均可看作是甲烷脫氫反應和氫氧化反應的耦合,而在芳構化過程中引入CO2或SO2等弱氧化劑,也是芳構化和“清碳、除氫”反應的耦合。這種體系既涉及不同反應之間能量傳遞過程的耦合也涉及反應物-生成物之間的功能互補。然而,從反應耦合視角來研究甲烷脫氫轉化過程的工作還遠遠沒有深入,值得進行廣泛而深入的探討。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
