富氧條件下Ag(x)/ZSM-5催化劑CH4-SCR脫硝性能
2021-03-21 04:48:18張相俊
工業催化 2021年12期
張相俊
(中國石油化工股份有限公司濟南分公司,山東 濟南 250100)
隨著我國對氮氧化物減排要求越來越嚴格,開發環保高效SCR脫硝催化劑受到研究者廣泛關注[1-2]。甲烷具有廉價易得的優點,許多研究者[3-6]對甲烷選擇性催化還原(CH4-SCR)脫除NOx進行過研究。以ZSM-5[2,6]、Al2O3[1,7]等為載體,活性組分主要采用Ag[2,8]、Cu[9-10]、Pd[11]、Co[12-14]、Fe[15-18]等。對于負載Ag物種的催化劑,Ag在載體上的負載形式與其來源(不同形式的鹽)有關,在反應過程中難以確定分布狀態;載體性質、反應溫度、反應氣氛等也會影響催化劑脫硝性能[19-20]。Richter M等[10]通過浸漬法制備了2%Ag/Al2O3催化劑,指出其催化活性主要與Ag的氧化狀態有關,Ag的完全氧化為催化劑上NO2的生成過程提供較多活性位點。也有研究者[10,19]證明,負載Ag型催化劑表面的納米銀物種提供了更強的NOx吸附中心。在低溫時,NOx在載體上的吸附物種會影響CH4活化[10,19-20],進而影響甲烷還原NO到N2的轉化率,隨著溫度升高,NO轉化率逐漸升高,同時甲烷逐漸發生氧化反應,降低了催化劑催化還原NO的選擇性[20]。
本文采用浸漬法制備Ag(x)/ZSM-5(x=0,3,6,9)催化劑,對催化活性組分負載量進行篩選,再對制備的催化劑進行表征,同時對催化劑上活性反應動態機制進行研究。
1 實驗部分
1.1 催化劑制備
采用低溫浸漬法制備Ag/ZSM-5催化劑。將適量硝酸銀(國藥集團化學試劑有限公司,分析純)置于一定量去離子水中,充分攪拌溶解后倒入ZSM-5載體(硅鋁比200),40 ℃恒溫攪拌4 h,靜置過夜,瀝去上層溶液,然后在圓底燒瓶中60 ℃、真空下旋蒸至液體完全蒸干得到催化劑前驅體,將其置于電熱恒溫鼓風干燥箱中110 ℃干燥12 h,最后在馬弗爐800 ℃下焙燒4 h,篩分,得到不同粒徑的催化劑。標記為Ag(x)/ZSM-5(x=0,3,6,9),x為Ag質量分數(以載體質量為基準)。
1.2 催化劑表征
X射線衍射在日本島津XRD-7000型X射線衍射儀上進行。CuKα,工作電流30 mA、工作電壓40 kV,掃描速率2o·min-1,掃描步長0.02o,掃描范圍10o~70o。
X射線光電子能譜(XPS)測試在美國賽默飛世爾公司ESCALAB-250型電子能譜儀進行。光源為Mg-Kα,功率為120 W。
催化劑顆粒形貌采用日本HITACHI公司S-4800型掃描電子顯微鏡(SEM)觀察,并利用其自帶的EMAX對樣品進行元素分析。
在北京昆侖永泰科技有限公司微型反應裝置上對催化劑(40~60目,0.15 g)進行程序升溫吸附-脫附測試(TPD)和程序升溫反應測試(TPSR)。預處理階段:550 ℃下Ar氣(30 mL·min-1) 1h。吸附段:100 ℃氣體氛圍30 mL·min-1(NO、O2、NH3、CH4、CH4+NO、NO+O2,Ar氣為平衡氣,下同)1 h。NO+O2-TPSR反應段:100 ℃下在30 mL·min-1混合氣(10%NO+10%O2)中恒溫吸附1 h。NO-CH4+O2-TPSR反應段:100 ℃下在30 mL·min-110%NO中恒溫吸附1 h,之后再通入30mL·min-1混合氣(10%O2,10%CH4)。脫附段:30 mL·min-1Ar氣中以10 ℃·min-1升溫速率從100 ℃升至700 ℃,反應尾氣采用英國Hiden公司QIC-20氣體質譜儀在線檢測尾氣,裝置降溫為自然降溫。
1.3 催化劑活性評價
催化劑活性評價在固定床微型催化劑評價裝置(北京昆侖永泰科技有限公司)上進行,以內徑為8 mm的石英管為反應器。反應氣體組成(均為體積分數)為:NO 600 μL·L-1,CH4600 μL·L-1,O25%,平衡氣為Ar氣,氣體總流量為300 mL·min-1。20~40目催化劑顆粒0.4g,以4 ℃·min-1的升溫速率對反應爐進行加熱,反應尾氣通入氮氧化物分析儀(42C-high level NO-NO2-NOx analyzer,Thermo Electron)進行實時檢測。
2 結果與討論
2.1 催化劑CH4-SCR脫硝活性評價結果
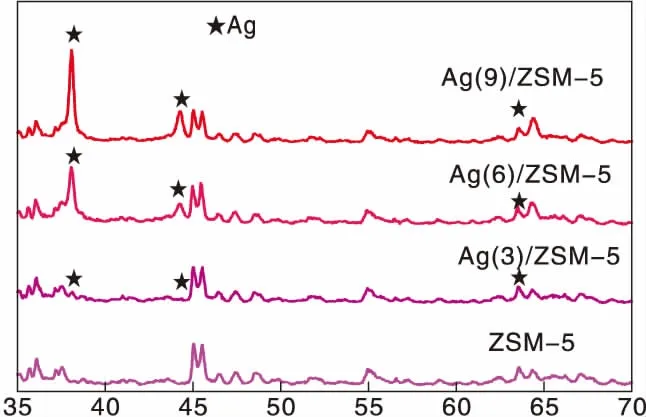
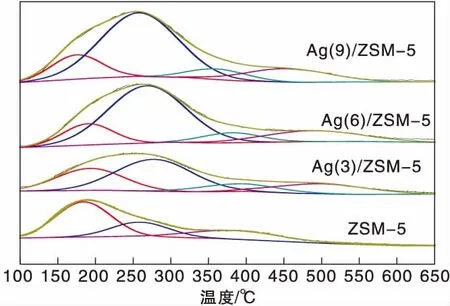
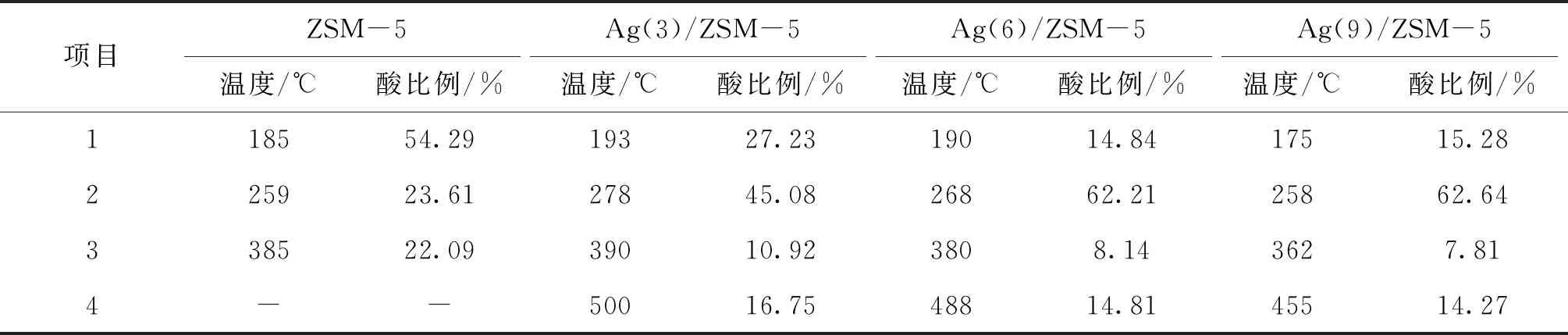
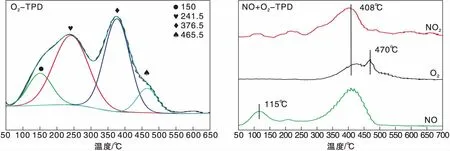
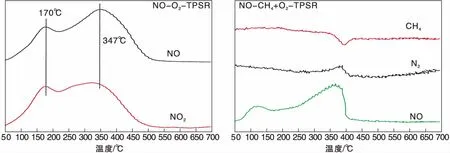
圖1為Ag(x)/ZSM-5(x=0,3,6,9)催化劑活性評價結果。從圖1可以看到,活性組分負載量對催化劑活性影響較大,NO轉化率隨Ag負載量增加逐漸提高,且高轉化率下的溫度向低溫方向移動。同時,由于CH4選擇性反應逐漸向氧化反應增強,催化還原反應活性隨之逐漸降低。從活性評價中可以得出:(1)在(280~475) ℃反應溫度區間,NO最高轉化率順序為Ag(3)/ZSM-5 圖1 Ag(x)/ZSM-5 (x=0,3,6,9)催化劑活性評價結果 圖2為Ag(x)/ZSM-5(x=0,3,6,9)催化劑的XRD圖。從圖2可以看出,活性組分的負載并沒有改變載體基本結構。Ag(9)/ZSM-5催化劑在2θ=38.06°,44.22°,64.34°出現金屬態的立方Ag晶相特征衍射峰,文獻[8]也有相關報道。根據謝樂公式計算催化劑Ag(9)/ZSM-5、Ag(6)/ZSM-5、Ag(3)/ZSM-5中Ag的晶粒粒徑分別為27.5 nm、30.6 nm和40.2 nm。當理論負載量增加時催化劑表面的Ag顆粒粒徑逐漸變小,主要原因是催化劑在制備過程中,隨著理論負載量的增加,在載體和Ag的作用下影響Ag在載體表面的分布,使其顆粒粒徑變小,為活性增加提供更大的比表面積,能夠起到提高其選擇性催化能力的作用。 圖2 Ag(x)/ZSM-5 (x=0,3,6,9)催化劑的XRD圖 Ag(x)/ZSM-5 (x=0,3,6,9)催化劑NH3吸附-脫附表征結果如圖3和表1所示。結合圖3和表1可以看出,(180~200)℃低溫脫附峰主要歸屬于催化劑外表面的酸性位(弱酸),主要是載體骨架外的部分裸露Ag陽離子[21]與NH3發生的化學配位吸附[5],即弱物理吸附。(400~500) ℃脫附峰對應B酸位上較強的NH3吸附[22]。隨著Ag負載量的增加,催化劑在低溫(小于200℃)下的NH3脫附量明顯增加、強度增強,但弱酸比例明顯下降,中強酸[NH3脫附溫度(200~400) ℃]比例逐漸增大,并隨溫度的升高脫附峰逐漸向低溫移動,Ag(3)/ZSM-5、Ag(6)/ZSM-5、Ag(9)/ZSM-5分別在500 ℃、488 ℃和455 ℃處形成新的強酸中心。說明負載Ag后弱酸強度降低,中強酸和強酸比例增大;負載量增加,脫硝活性提高,Ag(9)/ZSM-5催化活性最好;Ag在催化劑表面的分布影響Al離子分布,進而影響表面酸性位分布,Ag與載體上特定部位的結合或與分子篩骨架的相互作用而使酸性中心發生改變[22-23],使催化劑上強酸酸性增強,而酸性位溫度向低溫移動。 圖3 Ag(x)/ZSM-5 (x=0,3,6,9)催化劑的NH3-TPD譜圖 表1 Ag(x)/ZSM-5 (x=0,3,6,9)催化劑的NH3-TPD相關參數 圖4是載體ZSM-5(A1、A2)和催化劑Ag(9)/ZSM-5(B1,B2)掃描電子顯微鏡圖像。從圖4載體ZSM-5(A1,A2)照片可以看出,載體ZSM-5表面規整,具有較好的晶體形貌。B1、B2是Ag(9)/ZSM-5放大2萬倍和5萬倍的電鏡照片,從B1、B2可以看出引入Ag后,Ag形成細小顆粒分散在催化劑表面和孔道內,對載體表面形貌影響較大。 圖4 ZSM-5和Ag(9)/ZSM-5催化劑電子顯微鏡圖像 表2是Ag(9)/ZSM-5催化劑及載體ZSM-5的X射線能量色散譜圖數據。Ag(9)/ZSM-5經測試得出Ag實際負載量僅為5.63%,一方面是制備條件因素造成Ag損失;另一方面是負載Ag后造成載體表面物性改變。從元素含量分析知,負載Ag前后催化劑的Al和Si量降低,是部分Ag與載體上Al、Si發生交換所致,導致催化劑表面酸性、催化劑表面活性位等發生變化,進而導致催化劑選擇性催化還原NO性能的變化。另外,催化劑中C主要是催化劑吸附空氣中含碳化合物所致。 表2 ZSM-5和Ag(9)/ZSM-5催化劑X射線能譜數據 圖5是Ag(9)/ZSM-5催化劑的Ag3d光電子能譜圖。對譜圖進行分峰處理得到,Ag(9)/ZSM-5催化劑在374.6 eV 出現歸屬于金屬態Ag的振動特征峰,Ag3d5/2在367.5 eV是歸屬于Ag+電子結合能特征峰,而金屬態Ag的電子結合能則出現在368.3 eV[19-20]。表明Ag(9)/ZSM-5催化劑上Ag物種主要以Ag0為主,同時存在少量離子態Ag+。結合圖3結果,金屬Ag0在催化劑表面形成金屬簇,與載體作用形成有利于反應的活性位,進入載體骨架形成新的酸性中心[24],Ag+的形成是因為在催化劑高溫焙燒過程中存在少量Ag向分子篩孔道內遷移,這些Ag粒子遇到高溫下的B酸后與之結合形成金屬Ag離子。即高溫下Ag的遷移會導致催化劑上酸性位分布發生變化而影響催化劑的反應活性。 圖5 Ag (9)/ZSM-5催化劑Ag3d態光電子能譜圖 在CH4-SCR反應中,還原劑甲烷的活化是反應進行的關鍵環節,而富氧條件下NO在催化劑上吸附形成NOx物種如NO-、NO和NO+等[14,25]對甲烷的活化起重要作用[8]。圖6是Ag(9)/ZSM-5催化劑NO-TPD譜圖。圖6中NO的脫附峰在(100~500) ℃范圍內出現重疊現象,主要原因是催化劑表面能量分布不均勻,導致其對NO的吸附能力存在差異[19],即NO-TPD譜圖有幾個NO脫附峰則對應幾個不同的NO吸附中心。對NO-TPD譜圖進行分峰處理,得到峰值(脫附中心溫度)分別為175.5 ℃、345.5 ℃和437.5 ℃的3個脫附峰,峰面積占比分別為16.27%、46.60%和37.13%,即為催化劑在三個溫度下對NO的吸附百分比。結合催化劑的活性評價結果說明,SCR反應在(300~400) ℃內,NO更容易在Ag(9)/ZSM-5催化劑上發生吸附脫附作用,更有利于拓寬催化劑活性溫度窗口,為催化還原反應的進行提供更有利條件。 圖6 Ag(9)/ZSM-5催化劑NO-TPD譜圖 圖7是Ag(9)/ZSM-5催化劑CH4-TPD和CH4+NO-TPD譜圖。 圖7 Ag(9)/ZSM-5催化劑CH4-TPD和CH4+NO-TPD譜圖 由CH4-TPD譜圖可知,在(100~700)℃范圍內,沒有檢測到CH4脫附峰,說明CH4在實驗溫度下不能吸附在Ag(9)/ZSM-5催化劑上,或者吸附物種在此溫度范圍內很難脫附。在一定程度上能夠說明,由于Ag的載入,CH4在SCR中的作用機理是在催化劑表面迅速地參與反應而后又退出反應,或者是緊緊地吸附在催化劑表面充當反應活性位點而很難脫附下來。 對CH4+NO-TPD譜圖經分峰處理得到脫附中心溫度分別為133 ℃、228 ℃、333 ℃和423.5 ℃的低溫、中溫、高溫脫附峰,分峰所得到的各脫附峰面積占比分別為6.52%、10.48%、28.93%、54.07%。CH4分峰處理得中心溫度分別為195 ℃、275 ℃和403 ℃三個脫附峰,占比分別為7.93%、14.28%、77.79%。同單一NO物種吸附脫附譜圖(圖6)相比,NO和CH4競爭吸附時,NO低溫和高溫脫附峰溫度略有降低,中溫和高溫脫附峰所占比例略微下降。并且由CH4+NO-TPD譜圖可知,在(200~500) ℃溫度范圍內有CH4脫附峰出現,這與單一CH4脫附譜圖差別較大。原因是 NO存在時,NO在Ag(9)/ZSM-5催化劑上被吸附形成NOx吸附物種,導致催化劑表面理化性質發生變化,使催化劑增強對CH4的吸附[12-13],CH4在NOx吸附物種作用下易活化。對比圖6得到,競爭吸附也使NO脫附溫度降低。結合催化劑活性評價結果(圖1)得,NO在催化劑上的吸附能提高Ag(9)/ZSM-5對CH4吸附脫附性能,進而有利于催化劑對NO的選擇性催化還原。 圖8是Ag(9)/ZSM-5催化劑O2-TPD和NO+O2-TPD吸附-脫附譜圖。對O2-TPD譜圖分峰處理,在中心溫度分別為150 ℃、241.5 ℃、376.5 ℃和465.5 ℃的4個脫附峰,峰面積占比分別為12.69%、39.89%、39.22%和8.20%。4個脫附峰表明Ag(9)/ZSM-5催化劑上存在4種氧物種脫附中心。結合催化劑的活性評價結果,O2在Ag(9)/ZSM-5催化劑上發生脫附正好是催化劑活性劇增階段,NO轉化率在370 ℃比在200 ℃時增加了30個百分點,其在370 ℃處的脫附更有利于拓寬催化劑活性溫度窗口,并且O2脫附一直持續到500 ℃,這為催化還原反應中NO的氧化提供更多的O2吸附位。 圖8 Ag(9)/ZSM-5催化劑O2-TPD和NO+O2-TPD譜圖 NO+O2-TPD譜圖中,NO脫附中心溫度分別在115 ℃和408 ℃出現低溫和高溫兩個脫附峰,低溫脫附峰對應物理吸附的NO脫附產生。對比圖6可知,當O2存在時,同一NO的吸附物種在低溫與高溫處的脫附峰中心溫度均向低溫方向移動,且脫附量明顯降低。對比O2-TPD譜圖可知,各O2脫附峰中心溫度明顯升高,原因是NO與O2在競爭吸附的過程中形成了高價態的NOx中間體[25],由于NO與O2生成NO2是SCR反應的第一步[10,12,19-20],從圖中NO2譜圖可知,NO與O2競爭吸附過程中產生了少量NO2。 因此,在富氧條件下,催化劑對NO和O2的吸附作用影響CH4活化,O2與NO產生競爭吸附導致NO吸附向低溫方向移動且吸附量降低。同時,NO的吸附也對O2的脫附產生影響,使其向高溫方向移動并可能形成了高價態的不易脫附的NOx中間體。 由以上分析知,富氧條件下,NO2的生成是催化反應的首要步驟[10,20]。圖9是Ag(9)/ZSM-5催化劑的NO-O2-TPSR和NO-CH4+O2-TPSR譜圖。由NO-O2-TPSR譜圖可知,在富氧條件下,催化劑對NO的脫附依然存在170 ℃和347 ℃脫附峰,當有NO2生成時,NO的脫附隨之進行,其NO2信號趨勢基本與NO脫附峰一致,說明在O2存在下,吸附在催化劑上的NO及其物種發生氧化反應生成高價態NO2,這與文獻[14,26]報道一致。在(100~400)℃范圍內,隨著催化劑上吸附的NO及其相關吸附物種逐漸脫附,催化劑的CH4-SCR脫硝活性也逐漸降低。 圖9 Ag(9)/ZSM-5催化劑NO-O2-TPSR和NO-CH4+O2-TPSR譜圖 從NO-CH4+O2-TPSR譜圖可以看出,催化劑對NO的吸附脫附性能較好,在較低溫度下首先出現NO的脫附峰(125℃),驗證了在催化劑上弱酸中心(圖3結果)對NO的吸附作用較弱,對比NO-O2-TPSR譜圖,在氧的存在下,催化劑上逐漸發生氧化反應生成NO2吸附在催化劑表面,而這種中間產物(NO2)在較高溫 [(300~400) ℃)]下容易在催化劑表面形成較強的活性中強酸酸性位,為發生較強的CH4-SCR活性做鋪墊,這與NO+O2-TPD、NH3-TPD表征結果相一致。同時,從譜圖也可以看到,溫度(300~400) ℃區間,在催化劑作用下,伴隨著CH4的消耗而逐漸生成了N2,當溫度繼續升高,CH4的活化作用轉變成自身的氧化反應而使催化劑失去活性,CH4-SCR活性逐漸減弱直至消失[14,26],這也與催化劑的活性評價結果相一致。 因此,結合圖6、圖7、圖8的表征結果得知,在有氧且高溫條件下,催化劑對NO的吸附作用受到約束,同時甲烷氧化反應逐漸增強,使其選擇性逐漸降低,從而導致催化反應活性逐漸降低,這與催化劑的CH4-SCR活性評價結果一致。 通過浸漬法制備了Ag(x)/ZSM-5(x=0,3,6,9)催化劑并應用于富氧條件下CH4選擇性催化還原NO的研究。結果表明,增加催化劑的中強酸酸性而減弱催化劑的強酸酸性更有利于催化反應的進行,催化劑上的良好酸性位分布為其對反應氣氛的吸附提供更好的表面特性。Ag(9)/ZSM-5比制備的其他催化劑脫除NOx轉化率更高,這種差異歸因于催化劑上Ag及其物種不同狀態影響了催化劑對反應氣氛的吸附-脫附性能與氧化還原能力,催化劑上Ag主要以單質Ag存在,并為催化過程提供活性位,Ag→Ag+還原過程為催化反應帶來更高電荷轉移速率,這都能為反應提供更好的條件,以保證催化劑在較低溫度下具有較高的反應活性。在相同反應條件下,Ag(9)/ZSM-5催化劑NO轉化率大于30%的溫度窗口為(315~435)℃,隨著還原劑CH4氧化反應的進行催化劑失去活性。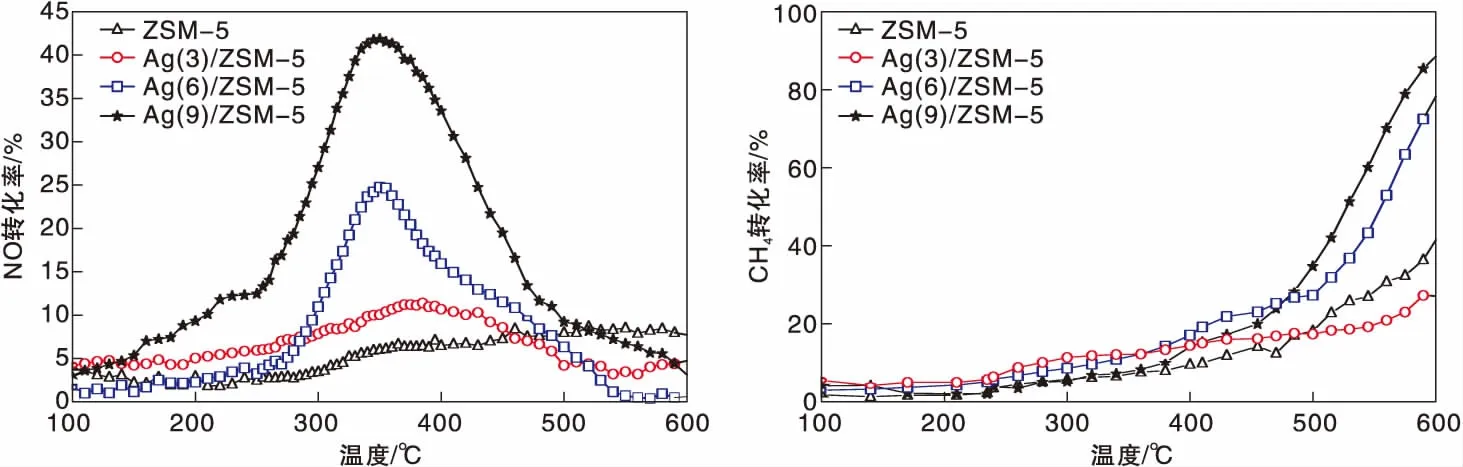
2.2 XRD
2.3 NH3-TPD
2.4 SEM及EMAX


2.5 XPS
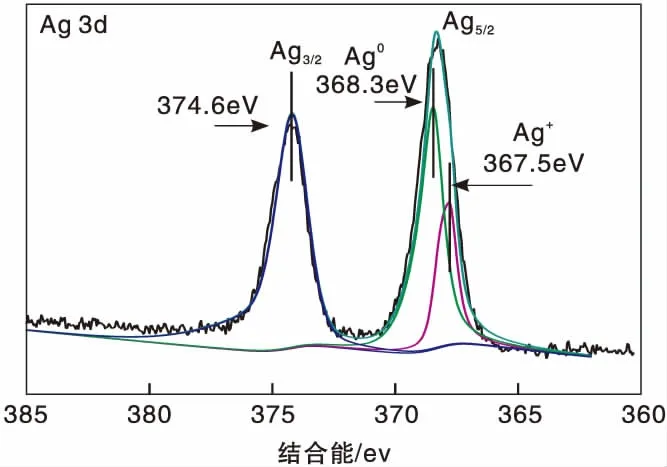
2.6 NO-TPD
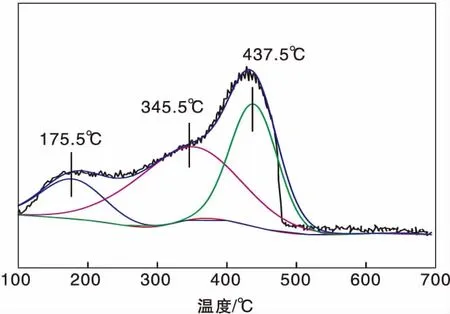
2.7 CH4-TPD和CH4+NO-TPD
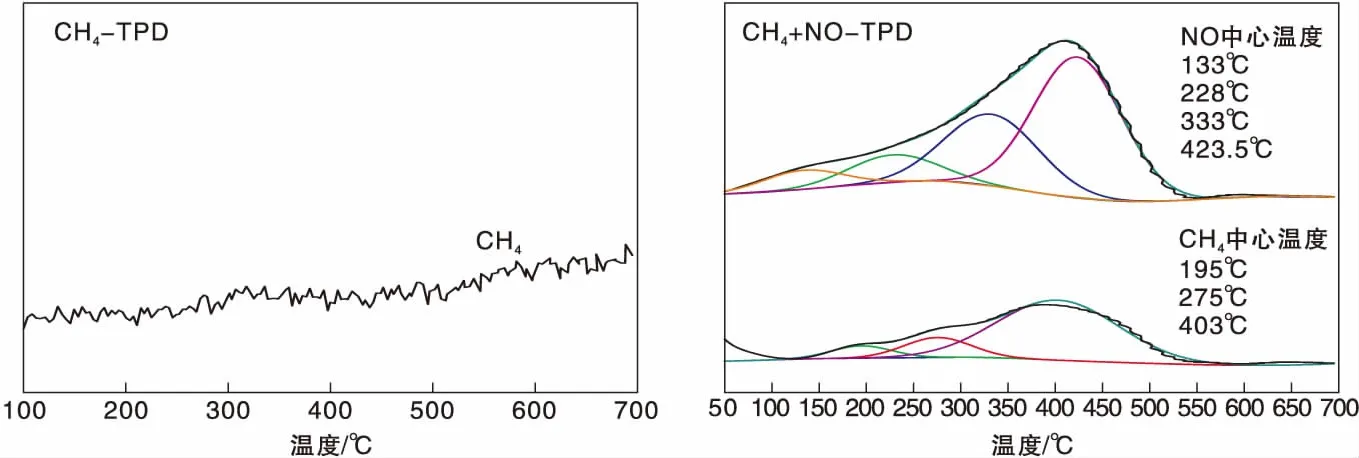
2.8 O2-TPD和NO+O2-TPD
2.9 NO-O2-TPSR和NO-CH4+O2-TPSR
3 結 論
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
