氮雜環丙烷[3+3]環加成反應研究進展
2021-04-02 02:11:40朱文慶張紫薇韓文勇
合成化學 2021年3期
朱文慶,張紫薇,韓文勇
(1.西安工程大學 環境與化學工程學院 陜西 西安 710048;2.遵義醫科大學 藥學院 貴州省生物催化與手性藥物合成重點實驗室 貴州 遵義 563006;3.遵義醫科大學 基礎藥理教育部重點實驗室暨特色民族藥教育部國際合作聯合實驗室 貴州 遵義 563006)
在新物質的合成過程中,雜環化合物扮演著極其重要的角色,是現代藥物分子結構中的重要組成。雜環類藥物因其雜環結構的特殊性而廣泛受到從事化學、藥學和生命科學研究科學家的高度關注,比如藥物分子中雜環結構的存在不僅影響藥物分子與受體之間的相互作用,而且可以降低藥物分子的親油性,從而有利于提高藥物分子的溶解度[1]。因此,如何快速并高效地進行雜環化合物的多樣性合成,一直以來都是有機化學家研究的熱點。值得注意的是,在含氮雜環化合物中,氮雜環丙烷作為最小且具有高度環張力的三元氮雜環化合物,是一類構建多種復雜氮雜環化合物非常重要的合成砌塊和中間體,被廣泛應用于許多天然產物和藥物分子合成中[2-4]。由于其結構的特殊性使其不僅可以與含碳、氮、氧、硫、鹵素等親核試劑發生開環反應生成開環產物[5-10],而且還可以與含烯基、炔基、羰基、氰基等多種底物發生[2+2]、[3+1]、[3+2]、[3+3]、[4+3]以及[5+2]等環加成反應,生成相應的多元氮雜環化合物[11-15]。本文將重點對氮雜環丙烷與多種不飽和鍵化合物的[3+3]反應進行系統地歸納與總結,以期為氮雜環丙烷在環加成反應中的應用提供理論參考,同時為促進創新藥物的研發與其新型合成技術的發展增添新的內容。
環加成技術的快速發展促使三亞甲基甲烷(TMM)金屬絡合物以及過渡金屬介導的TMM對不飽和鍵環加成反應引起了各研究學者的廣泛關注[16-18]。鑒于催化物種Pd-TMM的親核特性,Bambal和Kemmitt等[19]于1989年首次報道了2-[(三甲基硅基)甲基]-2-丙烯-1-乙酸酯1在Pd(PPh3)4作用下原位生成的三亞甲基甲烷鈀絡合物(Pd-TMM)與氮雜環丙烷2的[3+3]環加成反應(Scheme 1),以中等產率獲得了哌啶類化合物3。這一開創性的工作為后續Pd-TMM參與其它類型的環加成反應的發展奠定了基礎。

Scheme 1
基于Trost等[16,20,21]發現非堿性亞磷酸配體的加入可提高Pd-TMM配合物反應活性的認識,Harrity等[22]于2001年報道了在鈀催化下以P(OPr-i)3作配體,n-BuLi作還原劑,在溫和的條件下實現了原位生成的三亞甲基甲烷鈀絡合物(Pd-TMM)與手性氮雜環丙烷4的[3+3]環加成反應(Scheme 2)。在此工作基礎上,2003年該課題組[23]進一步探索了不同取代基的氮雜環丙烷6與Pd-TMM的[3+3]反應(Scheme 3)。發現氮雜環丙烷上的N-取代基R是該反應成功的關鍵,只有取代基R為4-甲苯磺酰基(Ts)和4-甲氧基苯磺酰基(PMBS)活化的氮雜環丙烷才能順利進行反應,而無芳基磺酰基活化的氮雜環丙烷不能參與此反應。同時,利用該方法學可將所得產物用于天然生物堿(-)-Pseudoconhydrine[22-23]和Indolizidine[24]的合成(Chart 1)。

Scheme 2

Scheme 3
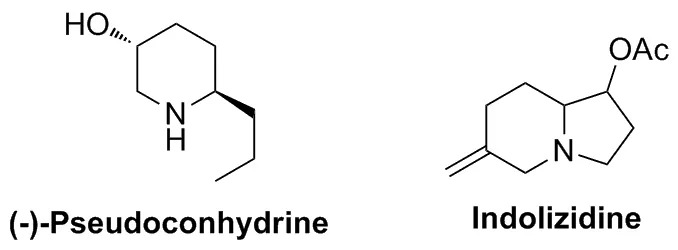
Chart 1
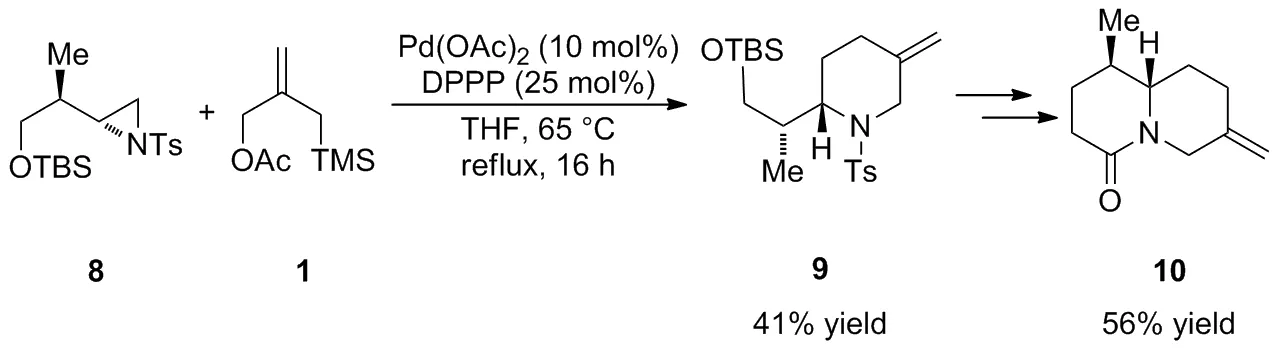
2003年,Harrity等[25]基于對Pd-TMM絡合物的反應特點的認識進一步采用鈀催化的[3+3]反應立體選擇性實現了哌啶衍生物9的合成(Scheme 4)。研究發現,單膦配體P(OPr-i)3會抑制Pd-TMM絡合物與N-對甲苯磺酰基氮雜環丙烷8的[3+3]環加成反應,而使用富電子的雙膦配體DPPP可以加速反應的進行,盡管以較低的產率(41%)獲得了環加成產物9,但經過后續的脫保護,Swern氧化和Wittig反應得到重要中間體10,可以進一步實現生物堿(-)-Deoxynupharidine1的合成(Chart 2)。
Scheme 4

Scheme 5
Scheme 6

Chart 2
鑒于上述Pd-TMM絡合物參與[3+3]環加成反應在哌啶類化合物合成中存在產率低的不足,尋求其他類型TMM等價物實現哌啶類化合物的高效合成是亟待解決的科學問題。2005年,該課題組[26]改進了哌啶類化合物的合成方法,通過2-甲基丙烯醇11在MgBr2作用下形成格式試劑與氮雜環丙烷8發生開環反應,隨后經分子內Mitsunobu反應,以87%的產率獲得了哌啶化合物9(Scheme 5)。同時高效地實現了中間體10的合成,進而實現生物堿(-)-Deoxynupharidine1,(-)-Castoramine2和(-)-Nupharolutine3的合成(Chart 2)。
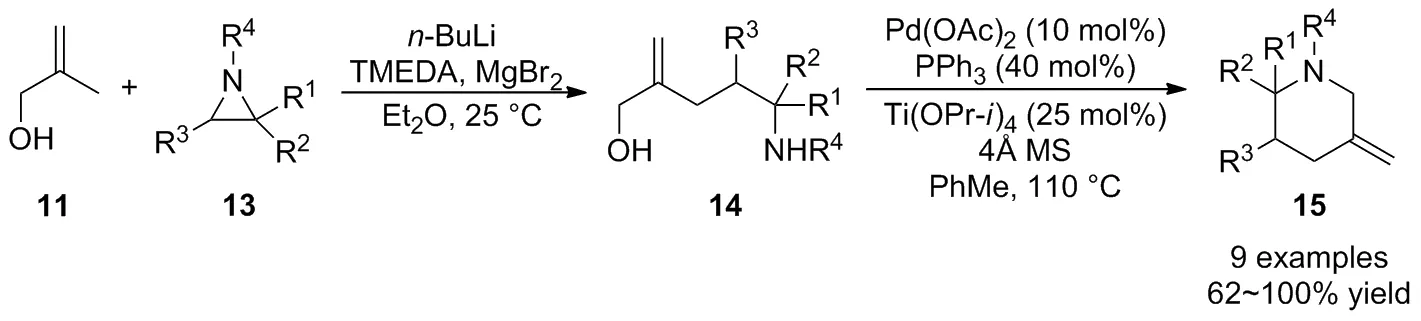
同年,該課題組[27]進一步通過2-甲基丙烯醇11在MgBr2作用下形成格式試劑與氮雜環丙烷13的開環反應,接著采用鈀催化的偶聯反應實現了哌啶類化合物15的高效構建(Scheme 6)。與先前開發的Pd-TMM介導的環加成反應相比,此法可用于反應活性較低的氮雜環丙烷,這極大地擴大了氮雜環丙烷參與[3+3]反應的底物范圍。
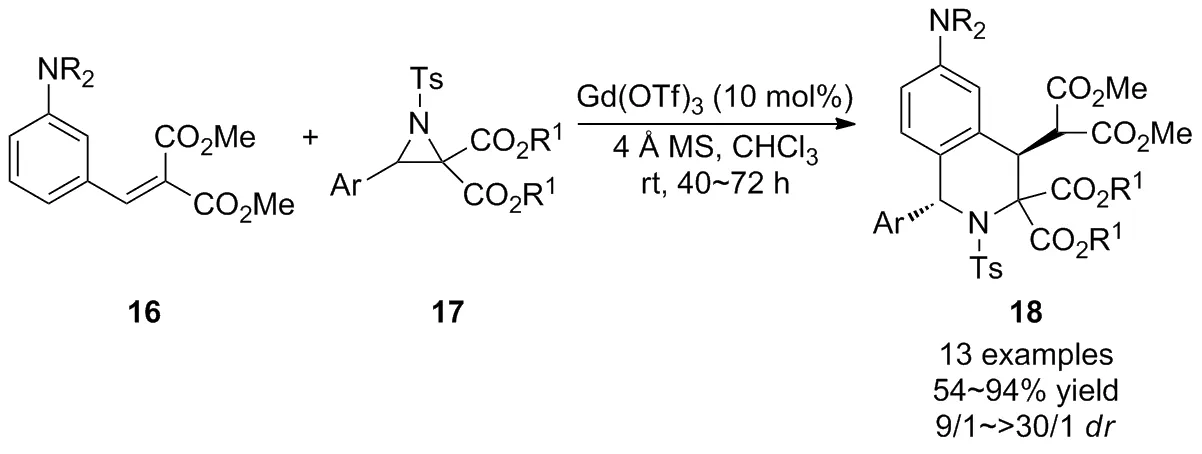
由于氮雜環丙烷具有較高的能量勢壘,關于其選擇性C—C鍵裂解生成偶氮甲亞胺葉立德進一步與親偶極分子發生[3+3]環加成反應的研究較少。2018年,Kim等[28]報道了Gd(OTf)3催化的N,N-二烷基-3-乙烯基苯胺16與D-A氮雜環丙烷17立體選擇性Friedel-Crafts/Michael串聯反應,通過D-A氮雜環丙烷選擇性C—C鍵裂解制備了高度官能化的四氫異喹啉衍生物18(Scheme 7)。基于上述研究成果,該課題組[29-30]利用相同的策略在溫和的條件下立體選擇性地獲得了具有1,4-碳立體中心的高官能化1,3,4-三取代的四氫異喹啉衍生物。
Scheme 7
Scheme 8
Scheme 9

Scheme 10
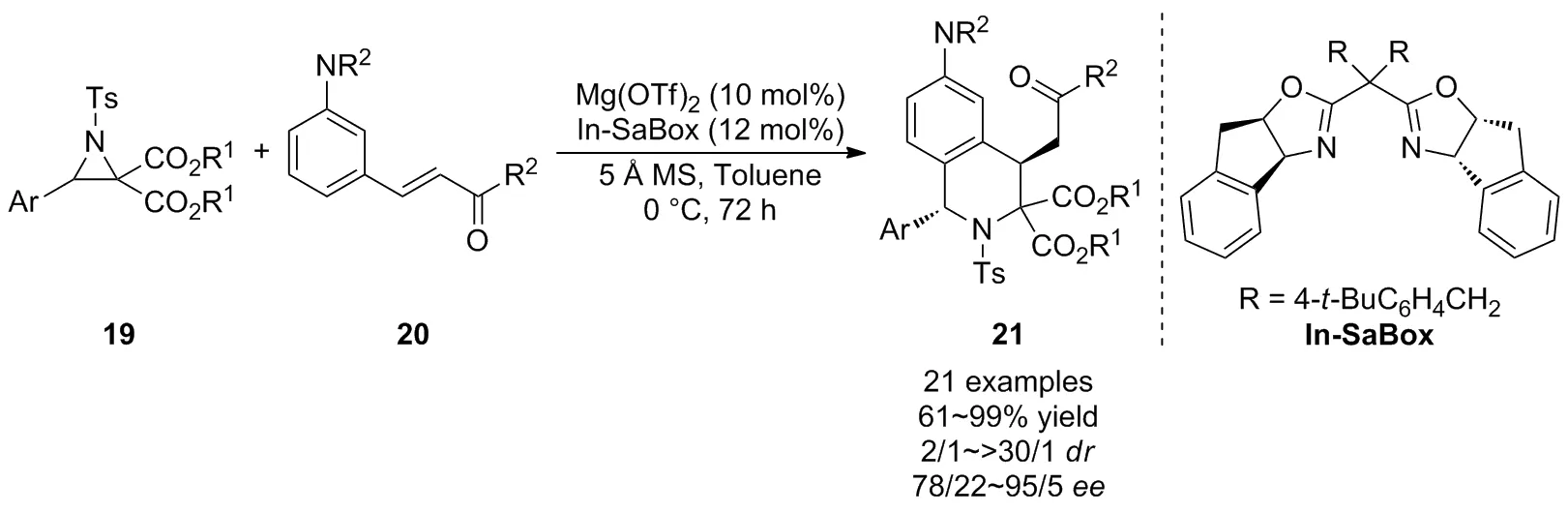
近年來,開發不對稱催化合成手性1,4-二取代四氫異喹啉衍生物的方法仍然具有挑戰性,2019年,Kim等[31]在前期工作的基礎上進一步報道了D-A氮雜環丙烷19與N,N-二烷基-3-乙烯基苯胺20的不對稱[3+3]環加成反應(Scheme 8)。以手性噁唑啉In-SaBox為配體,Mg(OTf)2為Lewis酸,在溫和條件下實現了產物的立體選擇性合成,以高達99%的產率,>30/1的非對映選擇性和95/5的對映選擇性獲得了一系列四氫異喹啉衍生物21。
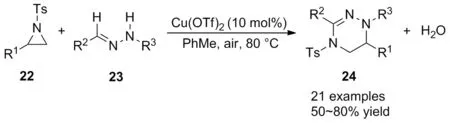
2009年,王彥廣和林旭峰[32]報道了在Cu(OTf)2催化下實現了氮雜環丙烷22與腙23的親核開環/分子間串聯氧化胺化反應(Scheme 9),以高達80%的產率合成了四氫三嗪類化合物24。該反應使用廉價的Cu(OTf)2為催化劑,空氣為氧化劑,溫和條件下實現[3+3]反應,水為唯一副產物。相比于Tsuge等[33]報道的氮雜環丙烷與腈亞胺合成四氫三嗪類化合物的策略,該方法更高效、綠色。
Scheme 11

Scheme 12

Scheme 13
1,2,4-噁二嗪烷是一類尚未開發的重要氮氧雜環化合物,其合成方法步驟繁瑣,且多用于產物的構象研究和藥物活性評價[34]。因此,Selander等[35]于2015年開發了一種InCl3催化氮雜環丙烷25與硝酮26的[3+3]環化反應(Scheme 10),以較高的產率和優良的選擇性合成了六元1,2,4-噁二嗪烷衍生物27。
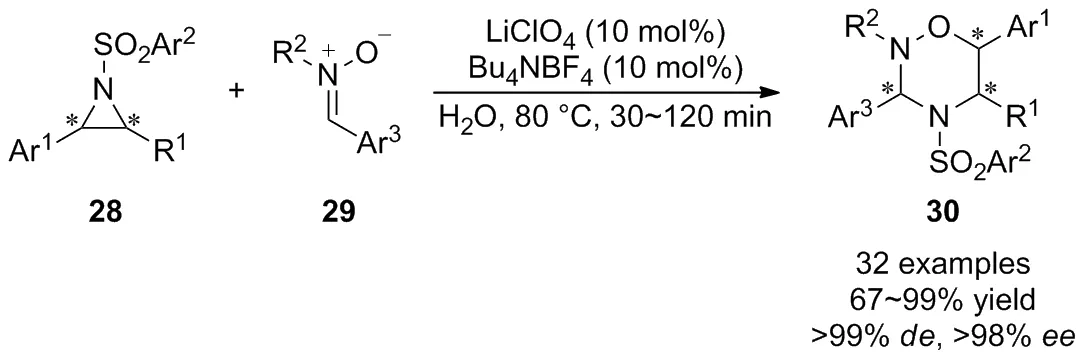
由于上述研究在反應步驟、底物范圍、立體選擇性控制等方面仍然存在著一定的局限性,開發一種新的合成策略實現手性1,2,4-噁二嗪烷衍生物的高效合成顯得非常必要。2017年,Ghorai等[36]首次在LiClO4/Bu4NBF4催化體系下通過活化的氮雜環丙烷28與硝酮29的[3+3]環化反應,實現了1,2,4-噁二嗪烷衍生物30的化學選擇性和立體選擇性合成(Scheme 11)。該反應成功地克服了氮雜環丙烷的競爭性氧化開環問題,反應條件溫和,底物范圍廣。
目前,關于氮雜環丙烷與雜原子的開環反應已經得到了廣泛報道,而與含碳親核試劑的開環反應仍然研究較少。2011年,Ghorai等[37]報道了t-BuOK介導活化氮雜環丙烷31與芳基乙腈32的SN2型開環,隨后在鈀催化下實現分子內C—N鍵偶聯反應,高效合成了多取代四氫喹啉衍生物33(Scheme 12)。底物普適性研究表明,不管是芳基取代的氮雜環丙烷還是烷基取代的氮雜環丙烷,均以優異的產率及立體選擇性獲得相應的四氫喹啉衍生物。作者還將該方法應用于多種外消旋和非外消旋的四氫喹啉衍生物的合成,產率高達99%,對映選擇性及非對映選擇性均可高達99%。
2018年,該課題組[38]進一步報道了路易斯酸BF3·OEt2介導的活化氮雜環丙烷34與α-異氰化物35的[3+3]環加成反應,經歷α-碳負離子的SN2型開環反應和分子內6-endo-dig環化反應,以優良的產率以及優異的立體選擇性獲得1,4,5,6-四氫嘧啶類化合物36(Scheme 13)。
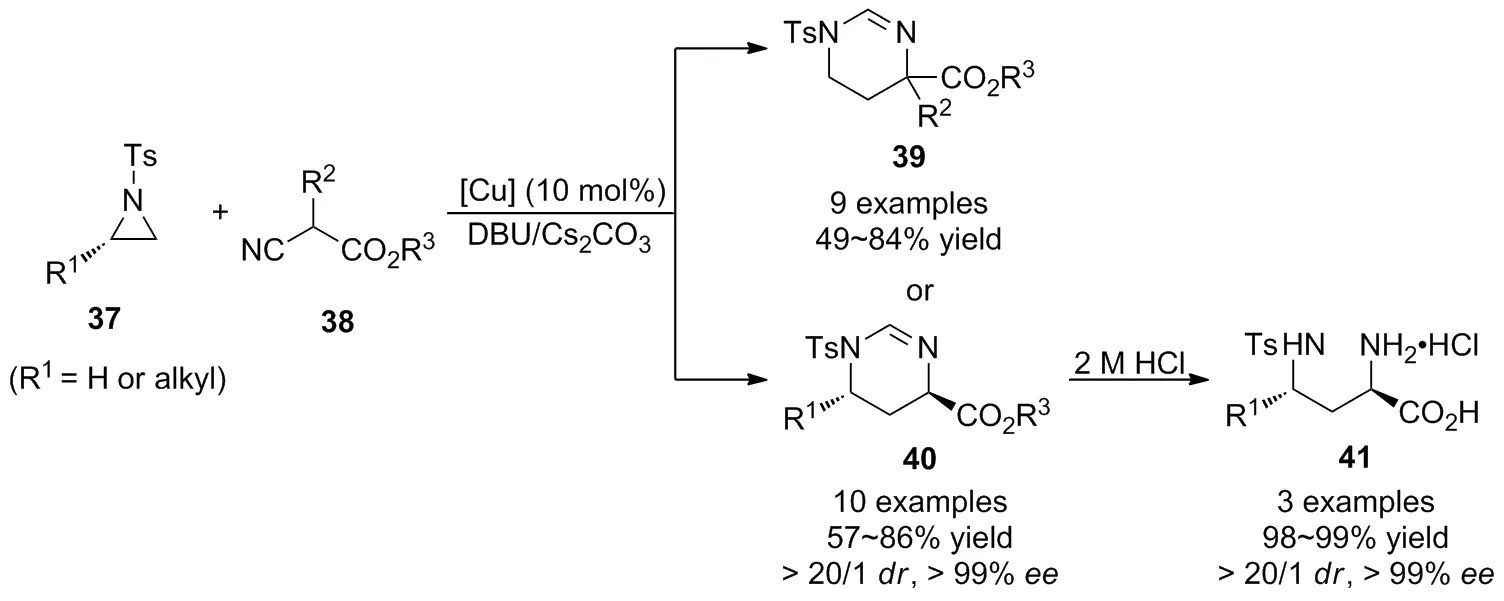
同年,趙宇等[39]又進一步報道了銅催化氮雜環丙烷37與異氰基乙酸酯38的[3+3]環加成反應(Scheme 14),以優異的立體選擇性獲得了1,4,5,6-四氫嘧啶衍生物39。經底物普適性考察發現,烷基取代以及芳基取代的異氰基乙酸酯具有良好的官能團耐受性,但其產率隨著異氰基乙酸酯中酯基位阻的增大而降低。為實現產物的對映選擇性控制,作者還篩選了大量的手性膦配體,氮配體以及二胺類配體,但仍未獲得預期的結果。為解決該科學問題,作者以對映體純的單取代氮雜環丙烷為底物,通過手性誘導策略,基于發展的銅催化[3+3]環加成反應實現了二取代四氫嘧啶單一立體異構體40的合成,經過簡單的水解過程即可獲得重要中間體手性α,γ-二氨基酸41的合成。
Scheme 14
Scheme 15

Scheme 16
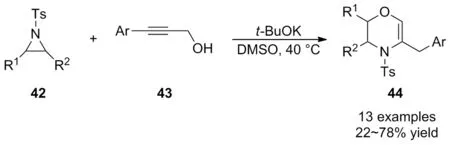
鑒于串聯反應在實現“一鍋煮”的多步反應中具有優越性,周永貴等[40]于2009年首次報道了溫和條件下t-BuOK介導氮雜環丙烷42與芳基炔丙醇43的開環/關環串聯反應(Scheme 15)。該反應意外的合成了羥嗪類化合物44,這一發現為六元羥嗪衍生物的合成提供了一種新方法。底物廣譜性表明,芐基取代的氮雜環丙烷參與反應所得產率偏低,原因可能是空間位阻造成的,而其它取代基的氮雜環丙烷和炔丙醇底物均具有良好的耐受性,可以中等至良好的產率獲得相應的目標產物。
同年,Sujit等[41]報道了[Ag(COD)2]PF6催化氮雜環丙烷45與炔丙基醇46的開環/環化串聯反應(Scheme 16),構建了多種六元或八元N,O-雜環化合物。對照實驗發現,Cs2CO3對環化反應具有促進作用。值得注意的是,使用非末端炔丙基醇或空間位阻較大的氮雜環丙烷反應時可以良好的產率獲得1,4-惡嗪衍生物47。
2009年,Kwon等[42]首次報道了膦作親核試劑介導氮雜環丙烷48與聯烯酸酯49的[3+3]環化反應(Scheme 17),進一步擴大了親核膦催化反應的范圍。該反應在溫和條件下進行,以良好至優異的產率及非對映選擇性獲得了多官能團化的四氫吡啶類化合物50。機理研究表明,反應經歷了兩步:分子內芳香親核取代反應以及SO2釋放的過程。

Scheme 17
Scheme 18
Scheme 19
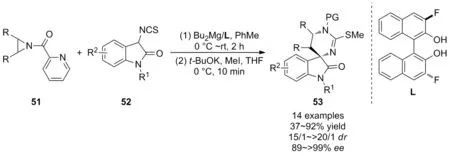
2015年,王銳等[43]首次報道了(R)-3,3′-F-BINOL/Mg絡合物催化的N-(2-吡啶基)氮雜環丙烷51與3-NCS氧化吲哚52的不對稱[3+3]反應(Scheme 18)。底物51和52在手性(R)-3,3′-F-BINOL/Mg催化劑作用下進行對映選擇性開環,進而在堿的作用下順利實現關環,以高達92%的產率,>20/1的dr值和>99%的ee值獲得了六元雜環化合物53。除此之外,開環的中間產物還可以進一步轉化為手性氨基酸、多肽和雙功能催化劑。
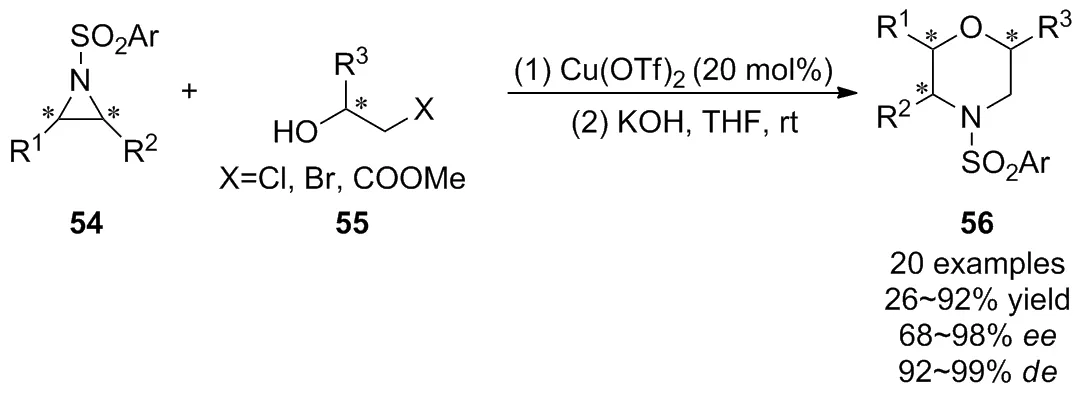
2009年,Ghorai等[44]報道了Lewis酸Cu(OTf)2催化氮雜環丙烷54與β-鹵代醇55進行SN2開環,接著通過“一鍋法”在室溫下進行分子內關環的高區域選擇性和立體選擇性的[3+3]反應,以優異的產率和對映選擇性構建了六元N,O-雜環化合物56(Scheme 19)。
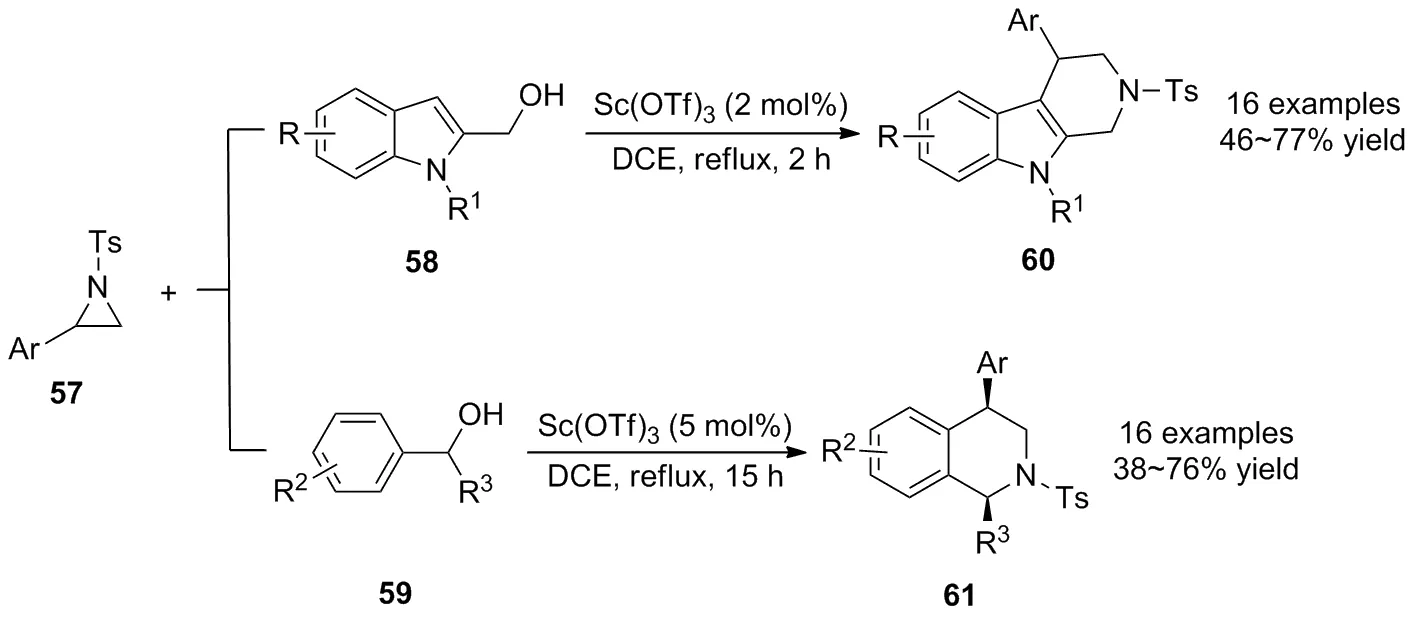
2013年,王紹武等[45]報道了Lewis酸Sc(OTf)3催化氮雜環丙烷57與N-烷基-2-吲哚甲醇類58和芐醇類59化合物的[3+3]環化反應(Scheme 20)。在溫和的條件下以簡單的原料獲得了四氫-β-咔啉衍生物60和四氫異喹啉衍生物61。底物廣譜性表明,對于不同取代基的氮雜環丙烷,均可獲得良好的收率。值得注意的是,芳基的存在有助于氮雜環丙烷的開環,而脂肪族的氮雜環丙烷則不發生反應;對于不同取代基的芐基醇,富電子的芐基醇與N-烷基-2-吲哚甲醇相比,其反應活性較低。最后,作者提出了該反應可能的機理,涉及到芐基醇與氮雜環丙烷的親核開環,中間體分子內胺化及Friedel-Crafts烷基化反應過程,是對Pictet-Spengler反應的進一步補充。
Scheme 20

Scheme 21
Scheme 22
Scheme 23
2017年,Banerjee等[46]通過Lewis酸MgI2催化D-A氮雜環丙烷62與1,4-二硫-2,5-二醇63的[3+3]反應,在室溫下實現了功能化噻嗪衍生物64的合成(Scheme 21)。底物普適性研究表明,含有吸電子取代基的氮雜環丙烷可快速轉化為目標產物,且對溴苯基取代的D-A氮雜環丙烷的反應活性高于鄰溴苯基取代的D-A氮雜環丙烷。重要的是,活性較低的苯基和萘基取代氮雜環丙烷也可以較好的產率獲得噻嗪類化合物;然而,含強給電子基取代的氮雜環丙烷穩定差,不能使環加成反應順利進行。有趣的是,作者在用含不同酯基的氮雜環丙烷考察酯基基團大小對反應轉化的影響發現,酯基的立體位阻對該反應的影響不大,均能以優異的產率獲得相應的環化產物。
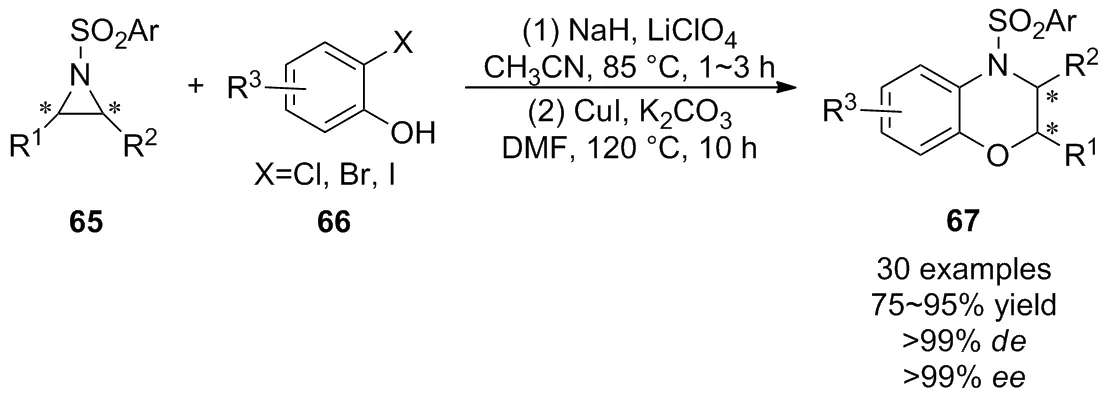
2018年,Ghorai等[47]開發了一種Lewis酸催化活化氮雜環丙烷65與2-鹵代酚66的SN2型開環反應,接著通過“一鍋法”實現了銅(I)催化分子內C—N鍵偶聯的[3+3]環化反應(Scheme 22),以良好至優秀的產率及優異的立體選擇性獲得了多種外消旋和非外消旋的二氫-1,4-苯并惡嗪衍生物67。
Scheme 24
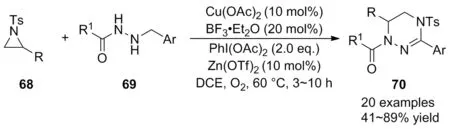
2019年,霍聰德等[48]在溫和的反應條件下首次實現了Cu(OAc)2催化氮雜環丙烷68與芐基肼類化合物69的氧化脫氫[3+3]環化反應,高效構建了功能化四氫-1,2,4-三嗪類化合物70(Scheme 23)。經底物普適性考察發現,芐基肼芳環上無論是給電子基團還是吸電子基團,都能以良好的產率得到相應的產物;多種N-Ts氮雜環丙烷苯環上的官能團也具有良好的耐受性。為說明該方法的實用性,產物還可通過脫保護和氧化脫氫過程轉化為更穩定的芳構化產物。
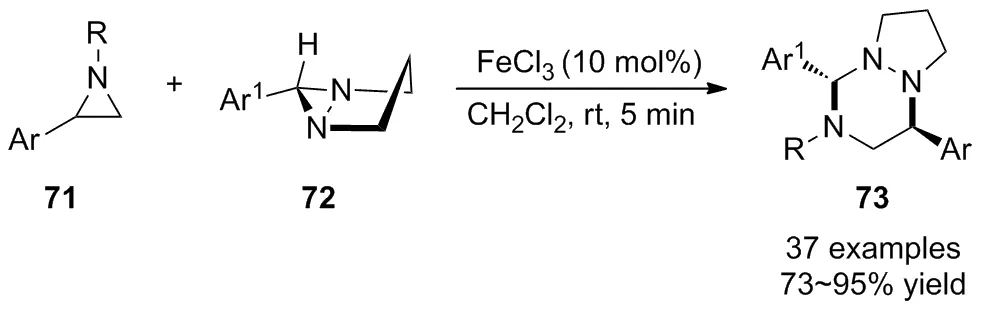
2020年,Sarkar等[49]報道了廉價FeCl3催化氮雜環丙烷71與二氮雜雙環[3.1.0]己烷72的立體選擇性[3+3]環化反應(Scheme 24),在室溫下高效合成了1,2,4-三嗪類化合物73。經研究發現,底物擴展到多種含吸電子基團或給電子基團取代的二氮雜雙環[3.1.0]己烷均表現出良好的官能團耐受性,含不同取代基的氮雜環丙烷均能參與此反應,以良好至優秀的產率得到相應的1,2,4-三嗪類化合物。作者還將該方法進一步應用到二氮雜雙環[3.1.0]己烷的環二聚體與N-烷基氮雜環丙烷底物的偶聯反應中,高產率地獲得了單一的非對映異構體。為了揭示其立體選擇性,用具有光學活性的氮雜環丙烷進行偶聯反應,結果表明該方法具有立體專一性,可獲得高光學純度的1,2,4-三嗪化合物。該報道為兩種不同三元環的環化反應提供了一種具有潛在應用價值的方法,進而為新型雜環化合物的高效合成提供了物質基礎。
綜上所述,經過化學研究工作者的不懈努力,實現了氮雜環丙烷與含C=C、C=N、C≡N、C≡C和C═·═C等基團化合物的[3+3]環加成反應,底物多樣性得到了進一步拓展,功能化多元氮雜環化合物的合成方法也得到了極大的改進與創新。但是目前仍然存在著底物范圍受限,反應活性較低及立體選擇性不理想等問題。隨著對含氮雜環化合物的不斷研究及其參與反應機理的探索,氮雜環丙烷與氧雜吖丙啶,環丙烷,亞硝基芳烴,N-亞磺酰基苯胺,環丙烯酮以及苯基烯酮等多種不同類型底物的[3+3]反應還有待進一步拓展,且構建高效的催化體系以實現氮雜環化合物的立體選擇性合成為藥物化學領域增添創新性成果顯得尤為重要。
