皮革及其制品中7種限用異噻唑啉酮類抗菌劑的氣相色譜/串聯質譜法測定
2021-04-14 09:17:22王成云褚乃清李泳濤沈雅蕾林君峰謝堂堂
西部皮革 2021年7期
王成云,褚乃清,李泳濤,沈雅蕾,林君峰,謝堂堂
(深圳海關工業品檢測技術中心,深圳 518067)
皮革及其制品中含有抗菌劑,以期提高產品的防霉變性能,異噻唑啉酮類抗菌劑作為一類廣譜、高效的抗菌劑,常用于皮革及其制品的防霉處理[1-4]。異噻唑啉酮類抗菌劑首先由美國Rohm&Haas公司取得專利權,開發出性能優良的成熟產品。作為一類非氧化型抗菌劑,異噻唑啉酮類抗菌劑對受體細胞膜和細胞壁具有很強的穿透能力,可穿透進入細胞內部,與含有巰基(-SH)的物質發生反應,生產S-S鍵,破壞細胞的再生繁殖,導致其死亡[5-6]。但是,大量研究結果表明,部分異噻唑啉酮類抗菌劑對人體和環境有害,對水生動物具有急性毒性[7-9],長期接觸時可引發接觸性皮炎、濕疹、皮膚灼傷、哮喘[10-22],職業性接觸時甚至可引起中毒性肝病死亡[23]。有鑒于此,各國立法限制使用部分異噻唑啉酮類抗菌劑[24-25]。目前限制使用的異噻唑啉酮類抗菌劑共有7種:4,5-二氯-2-正辛基-4-異噻唑啉-3-酮(DCOI)、2-甲基-4-異噻唑啉-3-酮(MI)、2-正辛基-4-異噻唑啉-3-酮(OI)、2-丁基-1,2-苯并異噻唑啉-3-酮(BBIT)、5-氯-2-甲基-4-異噻唑啉-3-酮(CMI)、2-甲基-1,2-苯并異噻唑啉-3-酮(MBIT)、1,2-苯并異噻唑啉-3-酮(BIT)。皮革及其制品中異噻唑啉酮類抗菌劑的測試已有文獻報道[26-29],但測試對象并未涵蓋全部7種限用異噻唑啉酮類抗菌劑。串聯質譜法采用多反應監測(MRM)模式進行定量,受基體干擾少,定量下限低[30-36]。本文采用超聲萃取-固相萃取柱凈化-氣相色譜/串聯質譜法技術,建立了1個氣質聯用分析方法,對皮革及其制品中7種異噻唑啉酮類抗菌劑進行了同時測定。
1 實驗部分
用色譜純甲醇(美國Fisher Scientific公司)將DCOI(純度99.8%、日本東京化成株式會社)、MBIT (純度98.0%,德國Dr.Ehrenstorfer GmbH公司)、MI(純度99.5%、美國Sigma-Aldrich公司)、BBIT(純度95.0%,德國Dr.Ehrenstorfer GmbH公司)、CMI (純度99.0%,德國Dr.Ehrenstorfer GmbH公司)、OI(美國純度99.9%,Sigma-Aldrich公司)、BIT(純度99.2%,美國Sigma-Aldrich公司)配制為混合標準儲備液,其中OI、BBIT、MBIT、BIT、CMI、DCOI、MI的質量濃度分別為19.19、20.49、20.75、38.40、39.20、39.67、40.23 g/mL。用甲醇將其逐級稀釋,配制混合標準工作液。
陽性樣品為市售黑色牛皮革,該樣品中同時檢出MI和CMI。將樣品剪成小塊,用Retch SM2000織物研磨儀(德國Retch公司)磨成粉末。稱取1.0 g粉末樣品,置于裝有25 mL甲醇的35 mL玻璃反應瓶中,用MJ-600超聲波清洗機(無錫市美極超聲設備有限公司)超聲萃取25 min,萃取溫度為40 ℃。過濾,用雞心瓶收集濾液。將雞心瓶轉移至RV10旋轉蒸發儀(廣州華馨科學儀器有限公司)中,在真空下旋轉蒸發至近干。將雞心瓶轉移至DCY-12氮吹儀(常州市萬合儀器制造有限公司)中,用干燥氮氣緩慢吹干,殘渣用2 mL甲醇溶解。用5 mL甲醇對Supelclean LC-Ph固相萃取柱(0.5 g/3 mL,美國supelco公司)進行預活化處理后,將所得溶液轉移到萃取柱中,用5 mL甲醇淋洗,收集流出液。將流出液在RV10旋轉蒸發儀中旋轉蒸發至近干,在DCY-12氮吹儀中用干燥氮氣緩慢吹干后,用1mL甲醇溶解殘留物[37-39],所得溶液用0.45 m濾膜(德國CNW Technologies公司)過濾后供GC/MS-MS分析用。
采用Shimazdu GCMS-TQ 8040MX氣質聯用儀(日本Shimazdu公司)進行測試。色譜分離在RXi-5MS(30 m0.25 mm0.25 m)色譜柱上進行,采用不分流進樣方式,進樣量為1L。程序升溫方式,初溫為90 ℃,以15 ℃/min的速度升至280 ℃,保持3 min。溶劑遲延為3min。進樣口溫度和傳輸線溫度分別為270 ℃、280 ℃。載氣為高純氮氣(純度>99.999%),載氣流速為1.1 mL/min。
質譜條件為:電子轟擊(EI)電離方式,電離能為70 eV,離子源溫度和四極桿溫度分別為210 ℃和150 ℃,多反應監測(MRM)條件見表1。
2 結果與討論
2.1 超聲萃取條件的優化
影響超聲萃取效率的主要因素有5個:萃取溫度、萃取溶劑種類、萃取時間、萃取方式、萃取溶劑體積[36-37,40-44]。首先考察萃取溫度(因素A)、萃取時間(因素B)和萃取溶劑體積(因素C)對萃取量的影響[41]。使用甲醇作為萃取溶劑對陽性樣品進行單次超聲萃取,只改變其中1個因素而另外2個因素維持不變,觀察萃取量的變化規律,結果發現,萃取量隨這3個因素變化而變化規律相同,當萃取溫度(萃取時間、萃取溶劑體積)增加時,萃取量逐漸增加并達到最大值,繼續增加時,萃取量反而有所下降。萃取量達到最大值時對應的萃取溫度、萃取時間、萃取溶劑體積分別為40 ℃、30 min、25 mL。設計了表2所示的三因素三水平正交實驗來考察這3個因素對萃取量的綜合影響,在表2給出的每個實驗條件下以甲醇為萃取溶劑對陽性樣品進行單次超聲萃取,測定其萃取量,結果見表3。該陽性樣品中同時檢出MI和CMI,對于MI和CMI,利用表3中的數據分別計算因素A、因素B和因素C的k值和極差,從而確定其優方案[45]。計算結果表明,對于MI和CMI,優方案均為A1B2C1。在此優方案給定的萃取條件下,以甲醇為萃取溶劑,對陽性樣品進行連續3次超聲萃取,測定每次的萃取量和總萃取量。對于MI,3次萃取量分別為1.95、0.04、0.00 mg/kg;對于CMI,3次萃取量分別為3.51、0.10、0.00 mg/kg。經1次萃取后,98.0%的MI和97.2%的CMI被萃取出來。可見,單次超聲萃取可將待測樣品中的目標分析物基本萃取完全。萃取溶劑種類是決定萃取效果主要的因素,由于皮革及其制品中含有大量的脂類物質,而脂類物質易溶于正己烷、丙酮、叔丁基甲醚、石油醚等弱極性溶劑,采用這些弱極性溶劑作為萃取溶劑時,會導致大量伴生雜質被萃取出來,嚴重干擾目標分析物的測試。為此選擇水、乙醇、甲醇、二氯甲烷、乙腈、乙酸乙酯等極性較強的溶劑作為萃取溶劑,對陽性樣品進行單次超聲萃取,MI和CMI的萃取量見表4。對于MI和CMI,甲醇的萃取效果均最好。因此,超聲萃取條件最終優化如下:采用單次萃取方式,以甲醇為萃取溶劑,萃取溫度為40 ℃,萃取時間為25 min,萃取溶劑體積為25 mL。
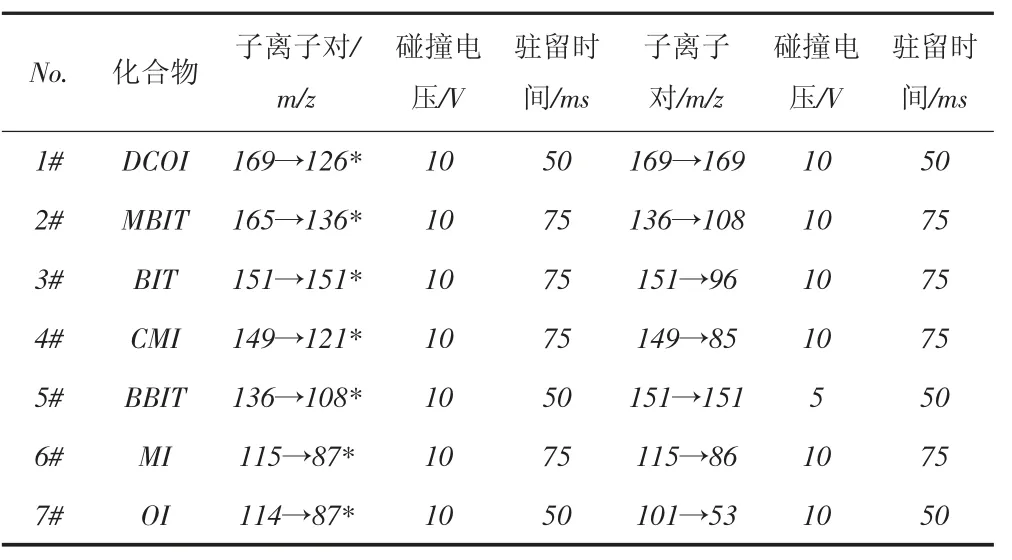
表1 MRM條件
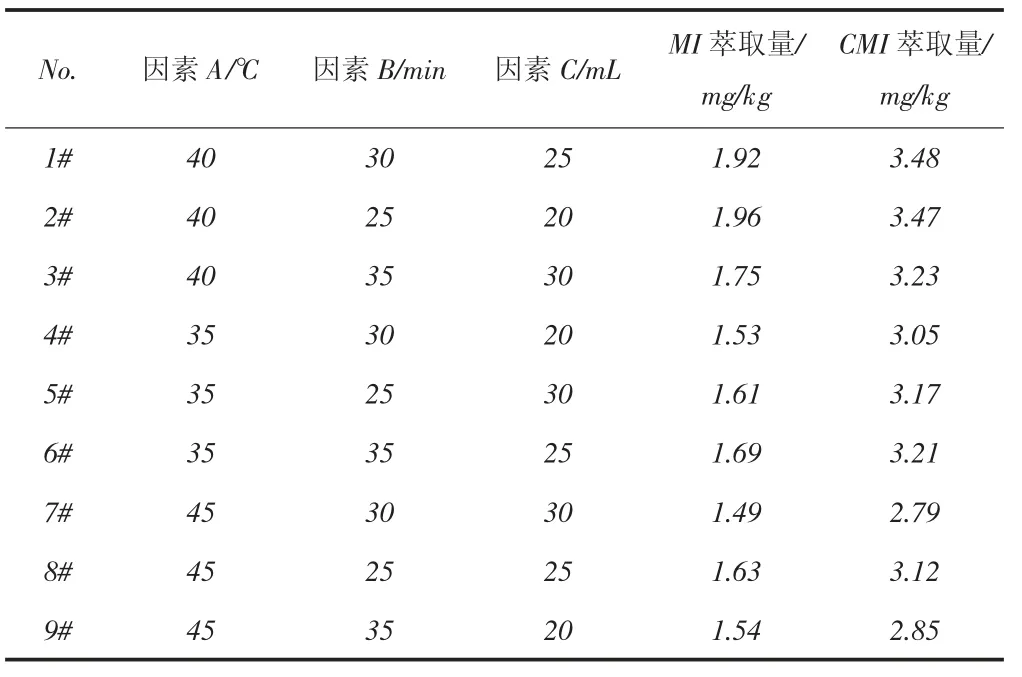
表2 正交實驗
2.2 凈化條件的優化
固相萃取柱(SPE)凈化技術基于液-固相色譜理論[46],利用選擇性吸附與選擇性洗脫的液相色譜法分離原理,采用選擇性吸附、選擇性洗脫的方式對樣品進行富集、分離和凈化。固相萃取柱的凈化效果主要取決于填料和容量,填料是色譜吸附劑,分為以硅膠為基質、以高聚物為基質、以無機材料為主三大類;吸附劑在吸附目標化合物時,也會吸附同類性質的雜質,容量過低時會導致回收率偏低。不同固相萃取柱對同一目標分析物的凈化效果各不相同[28-31]。經固相萃取柱凈化后,往往會損失部分目標分析物,也可能引入新的雜質。采用16種常見的固相萃取柱對混合標準儲備液進行凈化,測定各色譜峰面積,并與未經凈化處理的色譜峰面積進行比較,計算凈化后的回收率,結果見表5。從表5可知,經16種常用固相萃取柱凈化處理后,7種目標分析物均有所損失,其中1#柱Supelclean LC-Ph柱(0.5 g/3mL)損失最少,回收率均大于95%。對經這16種固相萃取柱凈化處理后的混合標準儲備液的譜圖進行比較,發現經1#柱處理后所得譜圖基本無雜質峰出現,基線平穩。因此最終選擇1#柱進行凈化處理。
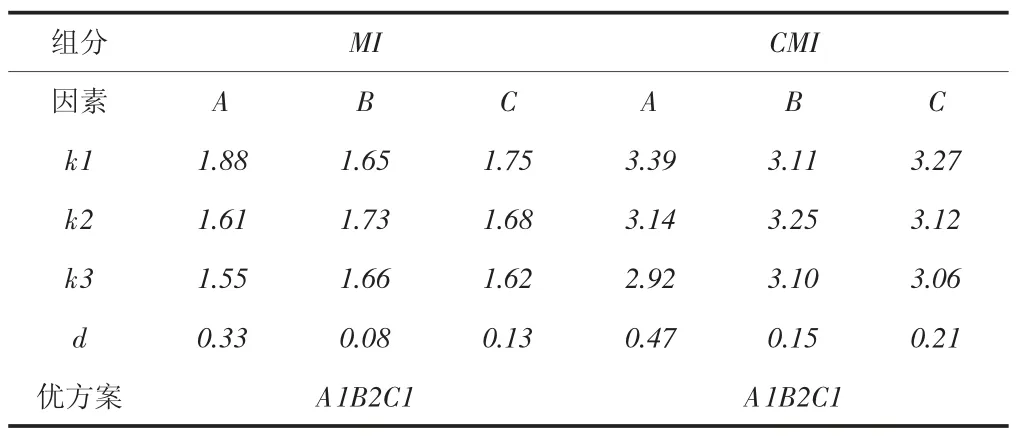
表3 正交實驗數據分析
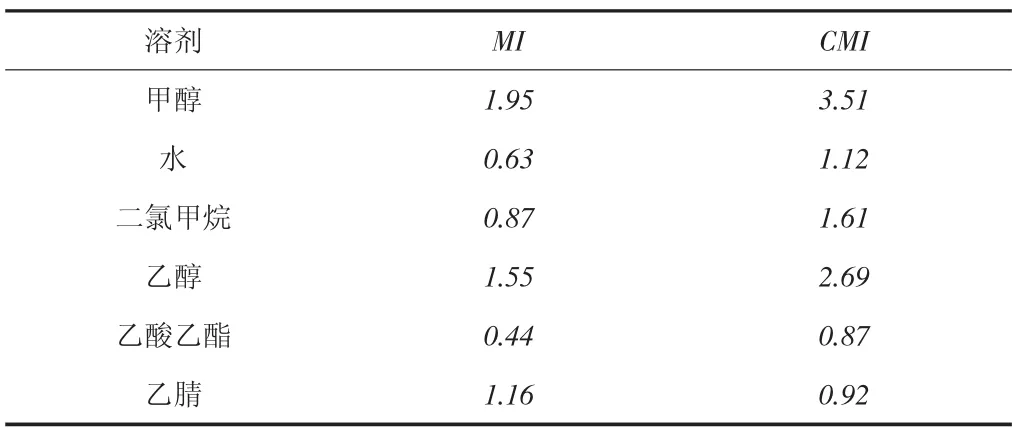
表4 不同溶劑的萃取效果/mg/kg
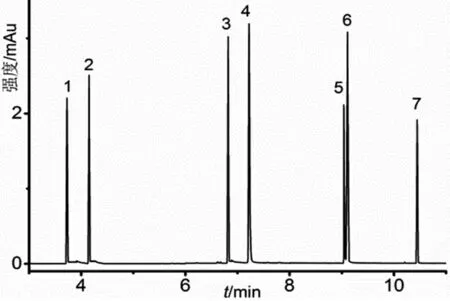
圖1 混合標準溶液的GC/MS-MS總離子流圖
2.3 分析條件的優化
混合物中各組分在色譜柱固定相和流動相之間分配系數各不相同,進行色譜分離時,各組分在固定相和流動相之間反復分配,從而使各組分獲得分離。影響色譜柱分離效果的主要因素是固定相的組成和色譜柱長度。考察DB-5HT (30 m 0.25 mm 0.10 m)、RXi-5MS (30 m 0.25 mm 0.25 m)、DB-17MS (30 m 0.25 mm 0.25 m)、DB-Wax (30 m 0.25 mm 0.25 m)、DB-5MS (60 m 0.25 mm 0.25 m)、DB-5MS (30 m 0.25 mm 0.25 m)、DB-624(30 m 0.25 mm4.10 m)、DB-Wax(60 m 0.25 mm 0.25 m)等8種常見色譜柱對混合標準溶液的分離效果,結果發現,RXi-5MS色譜柱的分離效果最好。改變升溫程序,觀察各組分的分離情況,在最佳升溫程序下,7種異噻唑啉酮類抗菌劑完全分離。
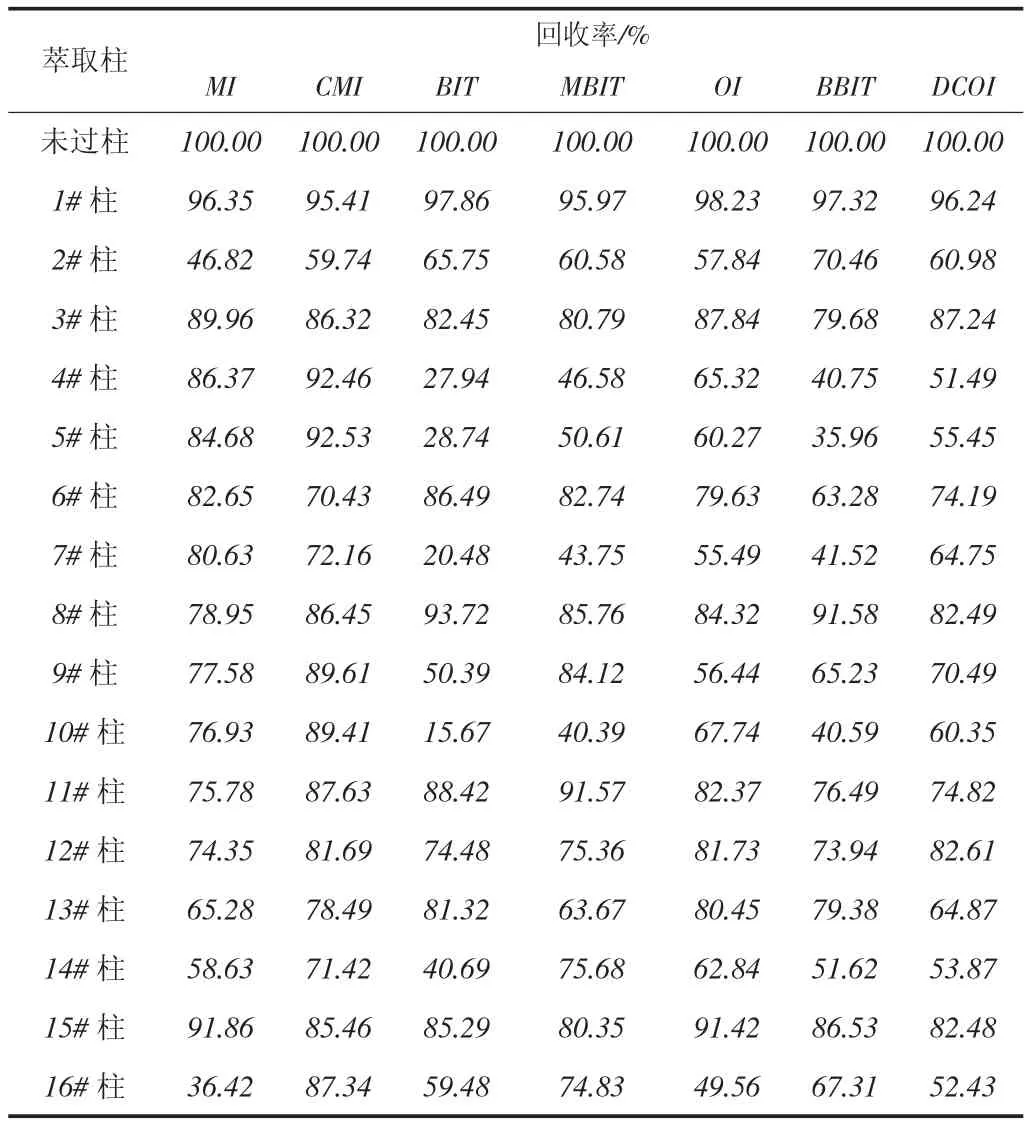
表5 經和未經固相萃取柱凈化后的回收率
對混合標準溶液進行全掃描質譜分析,得到總離子流圖,確定7種異噻唑啉酮類抗菌劑的保留時間和一級碎片離子。對于每種異噻唑啉酮類抗菌劑,選擇強度較高的2~3個一級碎片離子作為母離子,對其進行轟擊,產生二級碎片離子,所用碰撞電壓為15V。母離子與其產生的每個二級碎片離子可以組成一個子離子對。觀察每個子離子對的強度,對于每個母離子,選擇強度較大的2~3個子離子對用于MRM條件優化。采用分段掃描方式進行質譜分析,共設立3個時間段,分別為3.0~5.5 min、5.5~8.0 min、8.0~15.5 min。每個時間段內設立多個掃描通道,為了提高每個子離子對數據的采集質量,每個子離子對使用1個單獨的掃描通道。碰撞電壓從5V增加到50V,步長為5V,觀察每個子離子對的強度在不同碰撞電壓下的變化。對于每種異噻唑啉酮類抗菌劑,選擇強度最大的2個子離子對用于定性定量分析,其中強度最大的子離子對用于定量分析,這2個子離子對的碰撞電壓就是優化碰撞電壓。表1給出了7種異噻唑啉酮類抗菌劑的MRM條件,在此條件下對混合標準溶液進行測試,得到其MRM總離子流圖,如圖1所示。圖1中7種目標分析物譜峰完全分離,各譜峰峰形尖銳,對稱性好。
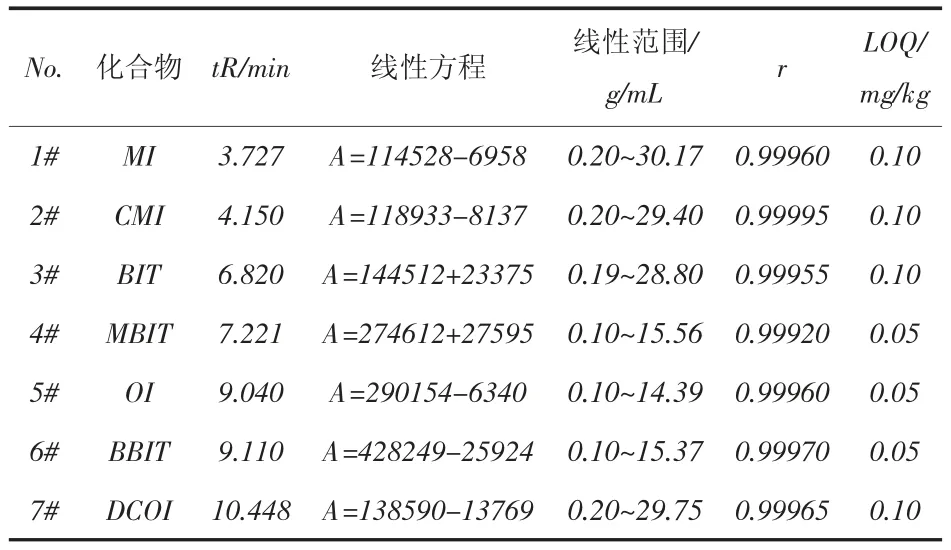
表6 線性關系和定量下限(LOQ)
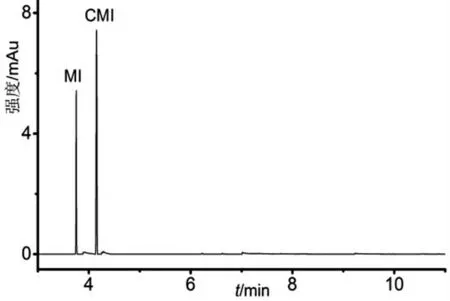
圖2 1個陽性樣品的GC/MS-MS圖
2.4 線性關系和定量下限
按1.3給出的分析條件對系列濃度的混合標準工作液進行測試,獲得混合標準工作液中各組分的峰面積(A),用峰面積對質量濃度()作圖,結果表明,在一定質量濃度()范圍內,峰面積(A)與質量濃度()之間存在良好的線性關系,線性相關系數大于0.9992。表6給出了7種異噻唑啉酮類抗菌劑的線性關系。在10倍信噪比(S/N=10)的條件下確定方法的定量下限,OI、BBIT、MBIT的定量下限均為0.05 mg/kg、BIT、CMI、DCOI、MI的定量下限均為0.10 mg/kg。
2.5 回收率和精密度
采用空白樣品加標回收的方法來確定方法的回收率和精密度[32],所用空白樣品分別為不含目標分析物的牛皮革、羊皮革和豬皮革,分別添加3個不同濃度水平的混合標準溶液,制備測試樣,每個添加濃度水平各制備9個測試樣。對各測試樣進行測試,測定各組分的含量,計算各組分的加標平均回收率和相對標準偏差(RSD,n=9)。方法的精密度用相對標準偏差(RSD,n=9)來表述,結果見表7。7種異噻唑啉酮類抗菌劑的加標平均回收率為80.25%~95.86%,方法精密度為2.84%~12.75%。
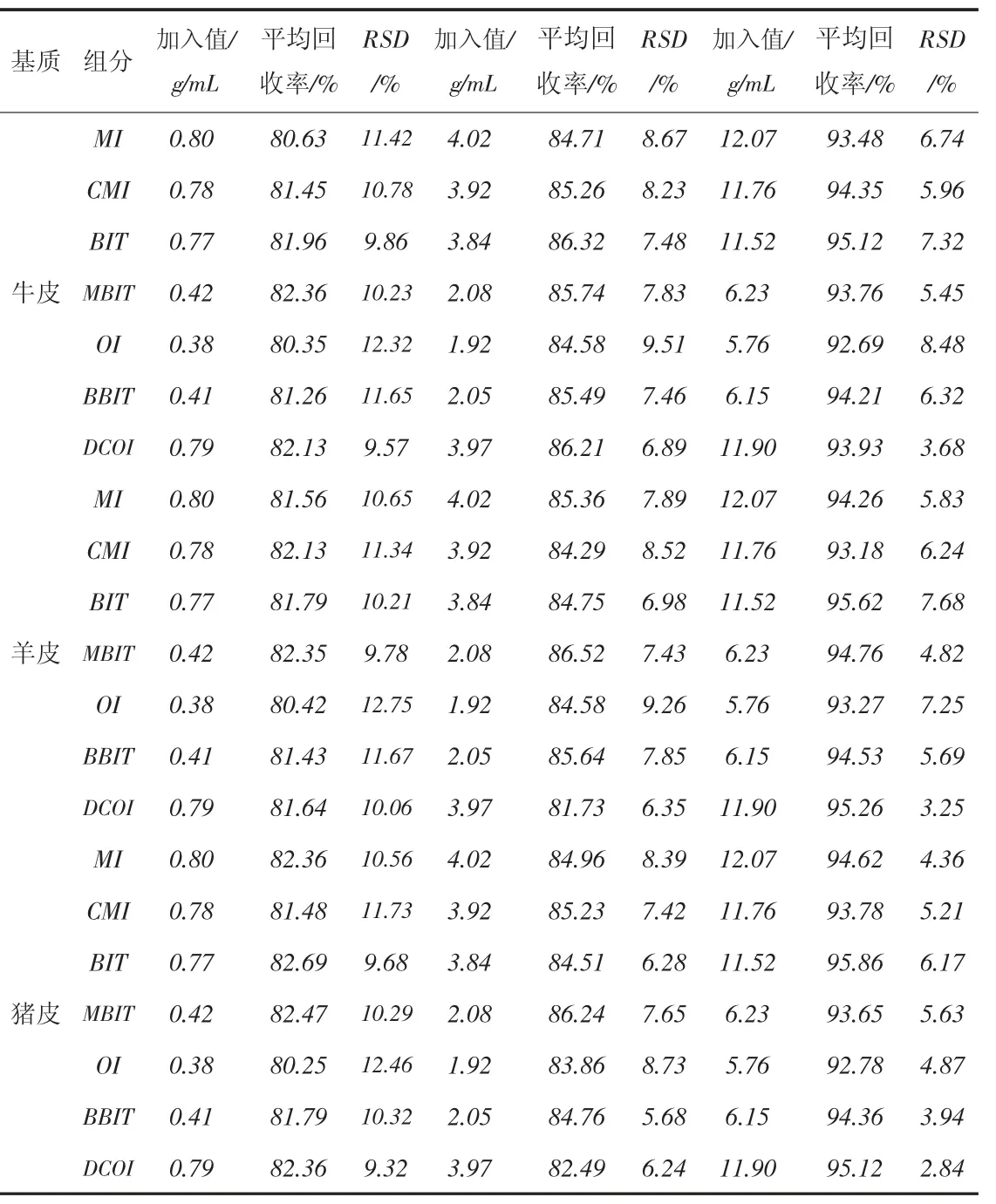
表7 回收率和精密度實驗
2.6 實際樣品測試
對市售皮革及皮革制品樣品中異噻唑啉酮類抗菌劑含量進行測試[28-29],測試樣品共32個,包括牛皮及牛皮制品樣品17個、羊皮及羊皮制品樣品8個、豬皮及豬皮制品樣品7個,結果在1個樣品中同時檢出MI和CMI,其余31個樣品中未檢出異噻唑啉酮類抗菌劑。陽性樣品為黑色牛皮革,圖2是其GC/MS-MS圖,該樣品中同時檢出MI和CMI,含量分別為1.95、3.51 mg/kg。
3 結論
建立了1個GC/MS-MS方法,對皮革及其制品中7種限用異噻唑啉酮類抗菌劑進行了同時測定。該方法利用超聲萃取技術提取待測樣品中的限用異噻唑啉酮類抗菌劑,提取物經固相萃取柱凈化后進行GC/MS-MS分析,外標法定量。該方法定量下限低至0.05~0.10 mg/kg,遠低于相關法規的限量要求,可完全滿足皮革及其制品中限用異噻唑啉酮類抗菌劑含量的日常檢測工作需要。采用該方法對市售皮革樣品進行測試,結果在部分樣品中檢出異噻唑啉酮抗菌劑。
