光度法測定凱氏氮存在的問題研究
2021-04-24 09:07:40周詩雨仇午丹趙晨晨
科學技術創新 2021年9期
關鍵詞:實驗
周詩雨 仇午丹 趙晨晨
(1、浙江人欣檢測研究院股份有限公司,浙江 寧波315000 2、浙江中環瑞藍科技發展有限公司,浙江 寧波315000)
隨著生態環境逐漸惡化,湖泊與水體富氧氧化現象普遍,凱氏氮是利用凱氏法對氮含量進行測量,可對水體富營養化程度進行有效評價。運用光度法測定凱氏氮,具有操作簡便、快速高效、試劑穩定等優勢,從而得到廣泛應用。在水樣采集完畢后開展實驗,在原有技術的基礎上加入特定的氫氧化鈉,繪制硼酸和氫氧化鈉體系校正曲線,使回收率得到極大提升,與實驗要求充分滿足。
1 凱氏氮測定方法
凱氏氮是利用凱氏法對氮含量進行測量,包括氨氮轉化為銨鹽后測定的有機氮化合物,該物質包含氨基酸、蛋白質、核算與尿素等,但不含有硝基化合物、疊氮化合物等等。在環境水中蘊含的氮以亞硝酸鹽氮、硝酸鹽氮為主,當水體受到污染后,導致大量有機氮融入其中,在微生物作用下分解,對水中溶解氧進行消耗,由此導致水質惡化。現階段,國內飲用水源調查是以凱氏氮為關鍵指標,如若該項指標超出設計標準,則說明水體受到污染,特別是對水中三種無機氮化合物一同測量時,有利于對水系自凈能力、污染程度進行分析,為湖泊與水庫富營養化評價提供有利指標[1]。
2 光度法測定凱氏氮實驗分析
2.1 儀器與試劑
該實驗主要利用型號為U-3410 紫外可見分光光度計、凱氏氮消解裝置、蒸餾裝置與分析天平。所用試劑包括硫酸鉀、氯化銨、硼酸、氫氧化鈉、六十酸鉀鈉,除氯化銨為優級純度之外,剩余均為分析純,實驗用水為無氨水。
2.2 實驗方法
在校正曲線繪制方面,準備8 個容量為50ml 的實驗瓶,每個瓶中加入濃度為12%、劑量為10.00ml 的硼酸溶液,與10.00ml 的氫氧化鈉溶液,搖晃均勻后,分別按照0、0.5、1.0、2.0、3.0、5.0、7.0、10.0ml 的氯化銨,再將1.0ml 的硫酸鉀鈉與1.5ml的納氏試劑加入其中,經過稀釋后搖晃均勻,沉淀10 分鐘后用比色皿測量吸光度,并繪制標準曲線。在樣品分析方面,選擇適量的河水樣品,將其放入容量為500ml 的凱氏瓶中,將10ml 的濃硫酸、2ml 的硫酸銅、6g 硫酸鉀與若干玻璃珠加入其中,搖晃均勻后放在通風柜中加熱,直至沸騰,將三氧化硫加入其中使溶液變得清澈,對熱源調整后保持微沸狀態30min,在常溫下冷卻后加入250ml 的水,攪拌均勻,當采集蒸餾液200ml 后停止操作[2]。
2.3 結果與討論
2.3.1 氫氧化鈉溶液加入量
采用特定的氯化銨與硼酸溶液量,分別加入不同體積的1mol 氫氧化鈉溶液,根據校正曲線實驗方式對吸光度進行測定,并對實驗現象細致觀察,結果如表1 所示。該實驗中硼酸用量為推薦值,在50ml 的容量瓶中將濃度為2%、劑量為10ml 的硼酸吸收液加入其中,使其與實際樣品條件一致;氯化鈉的用量為5.0ml。根據表1 可知,當氫氧化鈉溶液量過多或者過少時,膠體溶液均不夠穩定,吸光度與溶液加入量之間為正比關系,后者隨前者的增加而提升。對此,在實驗中應嚴格把控氫氧化鈉的用量。實驗表明,當該試劑用量為10ml 時,有色膠體溶液的穩定期最長,可將1mol 的氫氧化鈉溶液10ml 與硼酸中和,使膠體處于最佳穩定狀態。

表1 NaOH 加入量與吸光度關系
2.3.2 氫氧化鈉與硼酸物質量比例與用量
在只改變硼酸與物質量的比例,剩余條件固定的情況下,對膠體穩定性與吸光度的關系進行分析。當二者比例為0.3:1時,吸光度與膠體穩定期呈正比關系。在物質量比例固定不變下,其他溶液加入量變化對吸光度產生更大影響,實驗結果如表2。根據該表數據可知,當二者體積發生改變時,對膠體穩定性產生的影響較小,吸光度與二者體積具有正比關系。如若二者體積在0-8ml 之間時,與水樣吸收液添加量不符,本次實驗在創建校正曲線時,選用濃度為2%,劑量為10ml 的硼酸與10ml 的氫氧化鈉溶液作為校正溶液,用于縮短實際樣品測定期間的誤差。
2.3.3 校正與標準曲線對比
按照本文實驗方式繪制校正曲線,與標準曲線一同測定,根據測定結果可知,校正曲線斜率與標準曲線有所區別,因實際樣品測定中已經應用了硼酸與氫氧化鈉溶液。因此,為了使測定結果更貼近真實值,務必要利用與測定要求充分相符的校正曲線進行分析。在準確性檢測中,按照上述方式對某河水樣品進行分析,對凱氏氮回收率進行實驗。根據結果可知,利用校正曲線測定的凱氏氮含量結果更加可靠,樣品回收率在98-100%之間。在精密度檢測中,按照上述方式對某河水樣品進行分析,開展平行樣實驗,通過校正曲線法對樣品中凱氏氮含量進行測定。
根據實驗結果可知,平行實驗結果的標準偏差為0.36%,曲線法測定結果精密度更為理想。
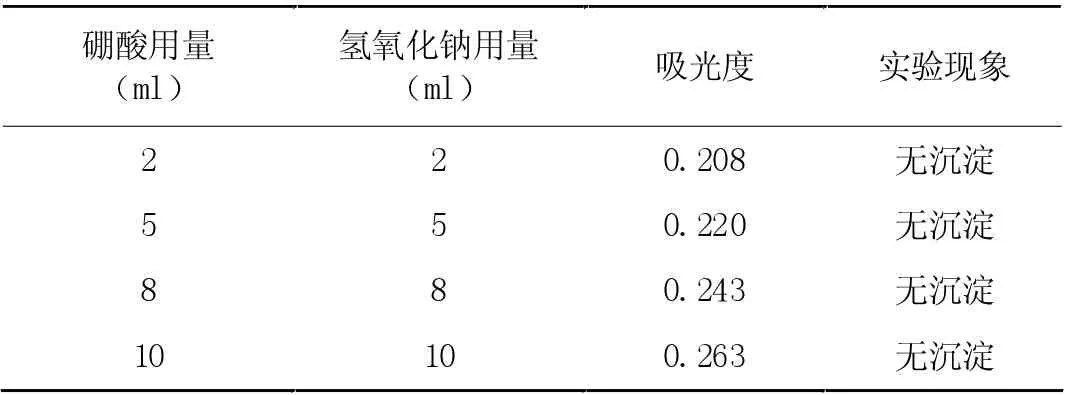
表2 兩種溶液用量影響
3 光度法測定凱氏氮問題的修正措施
3.1 取樣具有代表性
在天然水源中蘊含多種雜質,根據存在形態與顆粒大小的不同可分為不同形態,即膠體物、懸浮物與溶解物。其中,懸浮物的特點在于動水中以懸浮狀態存在,但在靜水中根據自重的不同上浮或者下沉。在地面水中無極懸浮物以泥沙為主,還包括礦物質廢渣、大粒徑粘土等等,此類物質自重較大,容易下沉;有機懸浮物的大小與來源不同,包括水草與小型浮游生物等等,主要源于污水、廢水之中;天然水中具有各類蛋白質、粘土膠體等等,前者是有機物分解階段產生,后者為無機顆粒,可提高水的渾濁度,導致水體變色,如在腐蝕物質影響下使水體變黃或者變為褐色。因此,在實驗選樣階段應確保均勻,具有代表性,免受主觀因素影響,且樣品采集后應及時分析,確保分析結果準確。
3.2 濃縮與消解同步開展
為了提高實驗結果準確性,濃縮與消解應同步開展,將操作流程進行簡化,由此節約更多時間,只要在上述操作中注重關鍵環節,針對相同樣品實施兩次消費與蒸餾,測得值重現性良好。針對相同地點在不同時間內兩次取水,采用滴定法對凱氏氮值進行測定,從中選出最佳指標制定最終實驗方案。此外,應加強消化階段的控制。以添加劑用量為主,國際標準為0.2g硫酸銅與6g 無水硫酸鉀,二者比例為1:30。如若添加量過多,消化液容易結塊,轉移難度增加;如若添加量較少,消化時間會延長,還會導致消化不完全。濃硫酸用量應根據樣品含水量、重量、蛋白質含量靈活調整。對于大體積、輕重量的產品來說,可適當增加濃硫酸用量,究其原因,此類樣品消化中濃硫酸對脫水、消化時的消耗量提升,如若添加濃硫酸量不足,很容易在碳化階段被燒干。在消化溫度方面,可通過循序加熱的方式,初始溫度為200-300℃,待試樣泡沫消失后逐漸提高溫度,達到360-600℃。當消化液定容后,在瓶口上加上漏斗,用少量蒸餾水多次洗滌燒瓶與漏斗,減少因誤差導致測定結果偏低的情況。在轉移完畢后禁止立即定容,而是要將其冷卻到室溫狀態后再定容。
3.3 做好蒸餾工作
實驗者應明確預蒸餾的重要意義,在蒸餾器正式應用之前,先將蒸餾水與NaOH 溶液混合起來實施預蒸餾,直到蒸出液無氨為止。儀器每次停用時間超過4 小時便會累積氨氣,再次使用時便要進行預蒸餾,確保管內壁附著的氨得到有效沖洗。同時,還應對蒸餾器嚴密性進行檢驗。在蒸餾作業中,為避免氨逃逸造成損失。在添加濃NaOH 溶液之前應將冷凝管與蒸餾瓶相連,然后緩慢的將濃堿液注入其中,馬上蒸餾。因氨具有較強的揮發性,快速蒸餾可將損失降到最低,使實驗結果更加準確可靠。最后,還應對蒸餾溫度進行嚴格控制。蒸餾裝置形式較多,大多采用水蒸氣蒸餾與直接蒸餾兩種形式,根據測定溶液含量與體積的不同,靈活選擇常量與半微量蒸餾。當上述兩種形式聯合應用時,務必要對溫度嚴格控制,如若蒸餾出現暴沸情況,很容易導致吸收液溫度提升,導致氨的大量損失,特別是以硼酸為吸收液的情況下,更要注重溫度合理。
3.4 加強滴定與測定控制
在實驗全過程中滴定屬于最后一個環節,其重要性不言而喻,應做好滴定控制工作,否則前面工作將會失去意義。在正式滴定前將溶液瓶搖晃均勻,將酸式滴定瓶沖洗2-3 次后加入鹽酸溶液,使內部氣泡排除,記錄刻度值后正式滴定。在滴定過程中應掌控好速度,接收液從淺綠色朝著無色轉變時放慢速度,逐一滴入鹽酸標液,到接收液變成淺紅色時結束,禁止滴量過多。在空白測定階段,額外選取兩個凱氏瓶,將相同劑量的試樣加入其中。根據實踐經驗可知,空白測定無需每次都開展,可隔一段時間開展一次。但是在更換實際、標準液時必須要開展空白測定。如若空白值超過規定標準,大多是因所用試劑級別不純、蒸餾水不純、玻璃器皿清潔不到位所致,需要對原因深入分析。
綜上所述,在對水體富營養化評價中,凱氏氮屬于重要指標之一。本文利用光度法對凱氏氮進行測定,充分發揮該方法試劑穩定、靈敏性強等特點。根據實驗結果表明,氫氧化鈉具有增敏效果,并要采用校正曲線保證結果準確性。為了修正實驗測定問題,應選取具有代表性的試樣,并加強對消化、蒸餾、滴定與測定等環節的控制,才可使實驗結果更加準確,更好的解決實際問題。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
