鼠傷寒沙門菌spvBC 基因編輯株的構建
2021-04-26 01:24:08左玲莉周麗婷吳興旗吳超逸吳淑燕
生物技術通報 2021年2期
左玲莉 周麗婷 吳興旗 吳超逸 吳淑燕
(1. 蘇州大學醫學部基礎醫學與生物科學學院,蘇州 215123;2. 蘇州高新區人民醫院醫學研究中心,蘇州 215129)
沙門菌(Salmonella)是一類經糞口途徑傳播的革蘭陰性菌,人和禽畜均可感染。2019 年加拿大六省沙門菌感染大暴發引起世界范圍內的關注[1]。根據宿主感染后的臨床表現,沙門菌可分為傷寒沙門菌(typhoidalSalmonella,TS)和非傷寒沙門菌(non-typhoidalSalmonella,NTS)兩大類,前者主要包括傷寒和副傷寒沙門菌。除上述沙門菌以外的血清學歸為NTS,在NTS 各血清型導致的全身感染中,鼠傷寒沙門菌(Salmonella typhimurium,S. typhimurium)是最常見的致病菌之一,約占65.2%[2]。免疫功能正常者感染鼠傷寒沙門菌多表現為胃腸炎型,免疫功能不全者則以敗血癥型和混合型多見[3]。我國感染性腹瀉病因中鼠傷寒沙門菌感染占23.53%[4]。與此同時,鼠傷寒沙門菌血清型的不斷變異和耐藥菌株的相繼出現為臨床治療增加了困難[5]。鼠傷寒沙門菌作為研究細菌致病機制與宿主相互作用的工具菌,已建立成熟的體內外模型[6]。因此,將其作為模式菌株,深入研究其致病機制并制定防控新策略具有重要的現實意義。
鼠傷寒沙門菌在與宿主的抗衡過程中,進化出一套精密的調控系統逃避宿主的免疫殺傷,其通過毒力島2(Salmonellapathogenicity island 2,SPI-2)依賴的Ⅲ型分泌系統(type Ⅲ secretion system,T3SS)分泌的效應蛋白形成沙門菌包裹囊泡(Salmonella-containing vacuole,SCV),為自身復制和繁殖提供有利場所[7]。鼠傷寒沙門菌常含約8 kb的沙門菌質粒毒力基因(Salmonellaplasmid virulence gene,spv),該基因與細菌的毒力表型密切相關。spv基因由spvR、spvA、spvB、spvC和spvD五個開放閱讀框組成[8]。其中spvB和spvC是研究最為廣泛的兩個毒力基因,與鼠傷寒沙門菌感染所致疾病的發生發展密切相關。spvB編碼的效應蛋白SpvB 羧基末端結構域具有ADP 核糖基轉移酶活性[9],能阻止肌動蛋白單體聚合,引起細胞骨架解聚[10-11]。本實驗室前期研究證明在鼠傷寒沙門菌感染進程中spvB可抑制宿主細胞自噬,加重損傷[12]。spvC編碼產物SpvC 具有磷酸蘇氨酸裂解酶活性,可特異性地使絲裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)信號傳導通路不可逆的去磷酸化失活[13]。文獻表明spvC可通過ERK 1/2 途徑抑制腸道早期炎癥反應,促進細菌全身播散[14]。本實驗室研究發現spvC影響焦亡與其抑制腸道炎癥促進細菌播散密切相關,但spvB與spvC的互作及其深入的致病機制尚未有文獻報道。
λRed 同源重組系統是一種基于同源重組原理,對細菌染色體DNA 直接修飾的基因編輯系統,可在原核生物中實現快速的基因敲除。該系統需要pKD46、pKD4 和pCP20 三種質粒。pKD46 是λRed同源重組系統的協助質粒,L-阿拉伯糖誘導后可表達exo、bet和gam3 個基因編碼的λ 噬菌體重組酶[15]。Exo 與Bet 引導線性片段與待敲除區域重組置換,Gam 抑制RecBCD 核酸外切酶的活性,使外源線性DNA 不至立即被降解[16]。pKD46 是溫度敏感復制子,30℃培養可表達重組酶,37℃培養質粒則丟失。pKD4 為同源打靶片段提供卡那霉素(Kan)抗性基因的模板,兩側含翻轉酶結合位點(Flipase recognition target,FRT),為重組轉化體提供篩選標志。pCP20 編輯的翻轉酶重組酶(Flipase recombination enzyme,FLP)可識別FRT 位點使之發生自身同源重組,從而消除一個FRT 位點及抗性基因[17]。pCP20 在42℃時誘導重組酶表達,同時質粒逐漸丟失。λRed 同源重組系統具有操作簡便,同源重組效率高,且對菌株生長整體無影響等優點[18-19]。
pBAD/g Ⅲ表達系統由araBDA 啟動子啟動,帶有g Ⅲ分泌信號和氨芐青霉素(Amp)抗性基因,L-阿拉伯糖能調控和誘導其表達,且表達產物羧基末端含有His 尾。這一標簽不影響目的蛋白的結構和功能,有利于蛋白的鑒定和篩選[20]。一般條件下,重組質粒的穩定性受細菌代數影響,研究表明利用pBAD/g Ⅲ原核表達載體構建的重組菌連續傳代30代后,質粒目的基因片段仍具有結構穩定性[21]。
鑒于此, 本研究利用λRed 重組系統和pBAD/g Ⅲ原核表達載體對spvBC進行基因編輯,構建鼠傷寒沙門菌質粒毒力基因spvBC敲除株和回補株,為深入探究鼠傷寒沙門菌致病機制及宿主的免疫應答提供可利用的工具。
1 材料與方法
1.1 材料
1.1.1 質粒和菌株 質粒pKD46、pKD4 和pCP20由普渡大學周道國教授惠贈。pBAD/g Ⅲ由江蘇大學黃新祥教授惠贈。鼠傷寒沙門菌標準株野生型SL1344 由南京農業大學楊倩教授惠贈。
1.1.2 主要試劑和儀器 T4 DNA Ligase、XhoI 及EcoR I 限制性內切酶購自日本TaKaRa 公司;Easy Taq DNA Polymersae 購自中國全式金生物技術有限公司;Kod-Plus-Neo 高保真PCR 酶購自日本ToYoBo公司;鼠抗His 抗體購自優寧維生物技術有限公司;鼠二抗購自R&D Systems 公司;質粒小提試劑盒、通用型DNA 純化回收試劑盒購自中國天根生化科技有限公司。PCR 擴增儀購自中國杭州朗基科學儀器有限公司;核酸電泳電泳系統購自中國上海天能科技有限公司;凝膠成像系統購自美國SYNGENE 公司;電轉化儀購自美國Bio-Rad 公司。
1.2 方法
1.2.1 引物設計 利用GenBank 公布的鼠傷寒沙門菌spvB(ID:13909717) 及spvC(ID:13909727)基因序列,設計敲除引物H1P1和H2P2。5'端分別為50 ntspvB基因上游和spvC基因下游同源序列,3'端為pKD4kan抗性基因(兩側含FRT 位點)的上下游引物序列。此外,在spvBC基因的外側設計一對鑒定引物P3和P4。該引物擴增產物中spvBC被kan替換后為1 747 bp,kan抗性基因消除后的敲除株中為356 bp。根據spvBC基因序列設計PCR 擴增引物spvBC-F(XhoI)和spvBC-R(EcoR I),并在引物5'端特異性酶切位點和保護堿基。引物序列見表1。
1.2.2 同源打靶片段的擴增 以pKD4 為模板,利用引物H1P1和H2P2PCR 擴增含spvBC同源臂及FRT 位點的kan抗性基因片段,PCR 反應體系:DNA 模板5 μL,引物H1P1和H2P2(10 μmol/L)各1.5 μL,Kod-Plus-Neo 高保真PCR 酶1 μL,10×PCR Buffer 5 μL,dNTPs 5 μL,Mg2+1 μL 去離子水30 μL。PCR 反應步驟:94℃ 5 min;94℃ 30 s,58℃ 30 s,68℃ 90 s,循環30 次;68℃ 7 min。利用DNA 純化回收試劑盒純化回收DNA 片段。

表1 實驗所用引物序列
1.2.3 感受態的制備及Red 重組酶的誘導表達 鼠傷寒沙門菌SL1344 37℃、220 r/min 振蕩培養過夜。按1∶50 轉接菌液于100 mL LB 液體培養基中,37℃、220 r/min 振蕩培養至OD600約0.6。將菌液冰浴20 min,在4℃條件下4 000 r/min 離心15 min,棄上清。加入冰浴的滅菌單蒸水10 mL 重懸,4 000 r/min 離心15 min,棄上清,重復洗滌3 次。再用冰浴的10%甘油10 mL 重懸,4 000 r/min 離心15 min,棄上清,洗滌1 次。最后加入冰浴的10%甘油500 μL 重懸,分裝后于-80℃保存備用。
將感受態細胞冰上解凍,無菌電轉杯冰上預冷。質粒pKD46 與感受態按1∶50 比例冰上混勻后進行電轉。電轉條件:電壓2.5 KV,電容25 μF,電阻300 Ω,電轉杯厚度2 mm。電轉后立即加入預熱到30℃的SOC 培養基混勻,30℃、220 r/min 振蕩培養3 h。離心去上清,收集菌體,均勻涂布于含100 μg/mL Amp 的瓊脂平板上,30℃培養過夜篩選陽性菌落。取新鮮單菌落接種于含3 mL 100 μg/mL Amp的LB 液體培養基,在終止培養前1 h 加入L-阿拉伯糖(終濃度30 mmol/L)誘導重組酶表達,再按照上述方式制備成含質粒pKD46 的感受態細胞。
1.2.4 同源打靶片段的電擊轉化及陽性克隆的篩選 取1 μL 純化后的同源打靶片段電轉入含質粒pKD46 的感受態細胞中,電轉條件同1.2.3。電轉后立即加入預熱的SOC 培養基混勻,30℃、220 r/min振蕩培養1 h 后37℃繼續培養2 h。離心去上清,收集菌體,均勻涂布于含50 μg/mL Kan 的瓊脂平板上,30℃培養過夜篩選陽性菌落。取單菌落利用引物P3、P4進行菌落PCR,鑒定完全重組的陽性菌落SL1344-ΔspvBC∷kan。
1.2.5 質粒pKD46 的消除 將陽性菌落涂布于Kan平板純化3 次后于42℃培養過夜,將獲得的單菌落分別涂布于含Amp 和Kan 的瓊脂平板上,37℃過夜培養。在Kan 平板上生長而在Amp 平板上不生長的KanRAmpS菌落即為消除pKD46 的陽性菌落。
1.2.6 卡那霉素抗性基因的消除 將消除質粒pKD46 的陽性菌落按方法1.2.3 制成感受態細胞備用。將質粒pCP20 按1.2.3 方法電轉入消除質粒pKD46 的感受態細胞中。產物涂布于Amp 平板,30℃培養過夜。將所得陽性克隆分別涂布于含Amp或Kan 的瓊脂平板上,30℃過夜培養。在含Kan 的平板上不生長而在含Amp 的平板上生長的AmpRKanS菌落即為消除Kan 抗性的陽性菌落SL1344-ΔspvBC∷FRT。
1.2.7 質粒pCP20 的消除 將上述單菌落涂普通平板于42℃過夜培養以消除質粒pCP20。所得陽性菌落同時涂普通平板、Amp 平板和Kan 平板,30℃培養過夜,普通平板生長而抗性平板不生長得菌落為成功消除kan片段和質粒pCP20 的敲除株。連續傳代3 次,利用引物P3、P4經PCR 鑒定spvBC基因敲除株SL1344-ΔspvBC。
1.2.8spvBC基因的擴增 以鼠傷寒沙門菌SL1344全基因組DNA 為模板,利用引物spvBC-F(XhoI)及spvBC-R(EcoR I)PCR 擴增含酶切位點的spvBC基因片段。PCR 反應體系同1.2.2。PCR 反應步驟:94℃ 5 min;94℃ 30 s,58℃ 30 s,68℃ 3 min,循環30 次;68℃ 7 min。利用DNA 純化回收試劑盒純化回收DNA 片段。
1.2.9 pBAD-spvBC重組質粒及回補株的構建 用限制性內切酶XhoI 和EcoR I 分別對上述PCR 擴增產物和質粒pBAD 雙酶切,酶切產物經瓊脂糖凝膠電泳鑒定,并切膠回收。將PCR 回收產物與載體質粒按體積比3∶1 混勻后經T4 DNA Ligase 在4℃連接過夜,最后將連接產物轉化至DH5α 感受態細胞中,篩選陽性克隆。利用引物spvBC-F(XhoI)和spvBC-R(EcoR I)進行菌落PCR 鑒定,根據鑒定結果測序,與GeneBank 數據庫中spvBC序列進行比對。將pBAD-spvBC重組質粒以電轉化的方式導入鼠傷寒沙門菌spvBC基因敲除株內,操作步驟同1.2.3,涂布于Amp 瓊脂平板篩選陽性克隆,連續傳代3 次后所得單菌落即為鼠傷寒沙門菌spvBC基因回補株SL1344-c-spvBC。
1.2.10 Western blot 檢測spvBC回補株SpvB 及SpvC蛋白的誘導表達 將回補株SL1344-c-spvBC接種到3 mL 含有Amp 的LB 液體培養基中,搖菌過夜培養,以SL1344-WT 和SL1344-ΔspvBC作為對照。取50 μL 菌液接種到5 mL 含Amp 的LB 液體培養基中繼續培養2 h,再分別加入1.3 mmol/L 和13 mmol/L L-阿拉伯糖誘導繼續培養3 h。將菌液4 000 rpm 離心5 min,棄上清,PBS 洗滌3 次。加入10 μL SDSPAGE 上樣緩沖液,混合均勻,將樣品置于100℃加熱5 min,-20℃保存待用。以SpvB 抗體檢測回補株SL1344-c-spvBC中蛋白SpvB 的表達,His 標簽抗體檢測回補株SpvC 蛋白(含His 標簽)的表達情況。取30 μg 樣品上樣進行SDS-PAGE 電泳(濃縮膠60 V,約40 min;分離膠120 V,約40 min)。將蛋白濕轉至PVDF 膜上(200 mA 恒流,60 min),置于搖床室溫封閉1 h。用抗體稀釋液稀釋鼠抗SpvB、His抗體(1∶1 000 稀釋),4℃孵育過夜。TBST 洗滌3次,每次10 min。抗體稀釋液稀釋山羊抗小鼠IgGHRP(1∶3 000 稀釋),室溫孵育1 h。TBST 洗滌3次,每次10 min。化學發光法顯影,檢測SpvB 及帶His 標簽的SpvC 蛋白表達水平。
2 結果
2.1 spvBC同源打靶片段的擴增
以pKD4 為模板,利用引物H1P1和H2P2PCR擴增spvBC同源打靶序列。該引物擴增含FRT 位點的kan片段為1 496 bp,加上兩端50 bp 的spvBC同源臂,PCR 擴增產物應為1 596 bp。瓊脂糖凝膠電泳顯示擴增產物大小與理論值相符,表明成功擴增出含spvBC同源臂和kan抗性的DNA 片段(圖1-A)。
2.2 spvBC同源打靶片段的轉化與重組
將純化后含同源臂的kanDNA 片段電轉入含質粒pKD46 的感受態細胞中,涂Kan 平板,30℃培養,篩選出陽性菌落。利用引物P3、P4進行菌落PCR鑒定,基因spvBC被kan替換后為1 747 bp。瓊脂糖凝膠電泳顯示擴增產物大小與理論值相符,表明kan抗性成功替換spvBC基因,即獲得完全重組菌SL1344-ΔspvBC∷kan(圖1-B)。
2.3 kan抗性基因的消除及spvBC敲除株的鑒定
將完全重組的陽性菌落SL1344-ΔspvBC∷kan涂布于Kan 平板純化3 次后于42℃培養過夜,篩選出KanRAmpS菌落即為消除pKD46 的陽性菌落。電轉入質粒pCP20,篩選出AmpRKanS菌落即為消除Kan 抗性的陽性菌落SL1344-ΔspvBC∷FRT。再將其42℃培養16 h,篩選出AmpSKanS菌落即為消除質粒pCP20 的spvBC基因敲除株SL1344-ΔspvBC。連續3 次傳代后,利用引物P3、P4進行PCR 鑒定,kan基因消除后敲除株中片段大小為356 bp。瓊脂糖凝膠電泳顯示擴增產物大小與理論值相符,表明成功構建spvBC基因敲除株SL1344-ΔspvBC(圖1-B)。
2.4 spvBC基因的擴增
以鼠傷寒沙門菌野生株SL1344 全基因組DNA為模板,利用引物spvBC-F(XhoI)及spvBC-R(EcoR I)PCR 擴增含酶切位點的spvBC基因片段,相應引物擴增的spvA、spvB、spvC和spvD基因片段作為對照。PCR 擴增目的產物片段大小應為2 789 bp。瓊脂糖凝膠電泳顯示擴增產物大小與理論值相符,表明成功擴增出含酶切位點得spvBC基因片段(圖1-C)。
2.5 pBAD-spvBC重組質粒的構建
spvBC基因片段和質粒pBAD 經XhoI 和EcoR I 雙酶切后切膠回收,回收產物連接轉化。隨機挑選單菌落,利用引物spvBC-F(XhoI)和spvBC-R(EcoR I)進行菌落PCR,10 號為鑒定出的陽性菌落(圖1-D)。將該菌落PCR 反應產物送測序,測序結果與GeneBank 數據庫中spvBC序列進行比對,結果表明pBAD-spvBC重組質粒的構建成功。
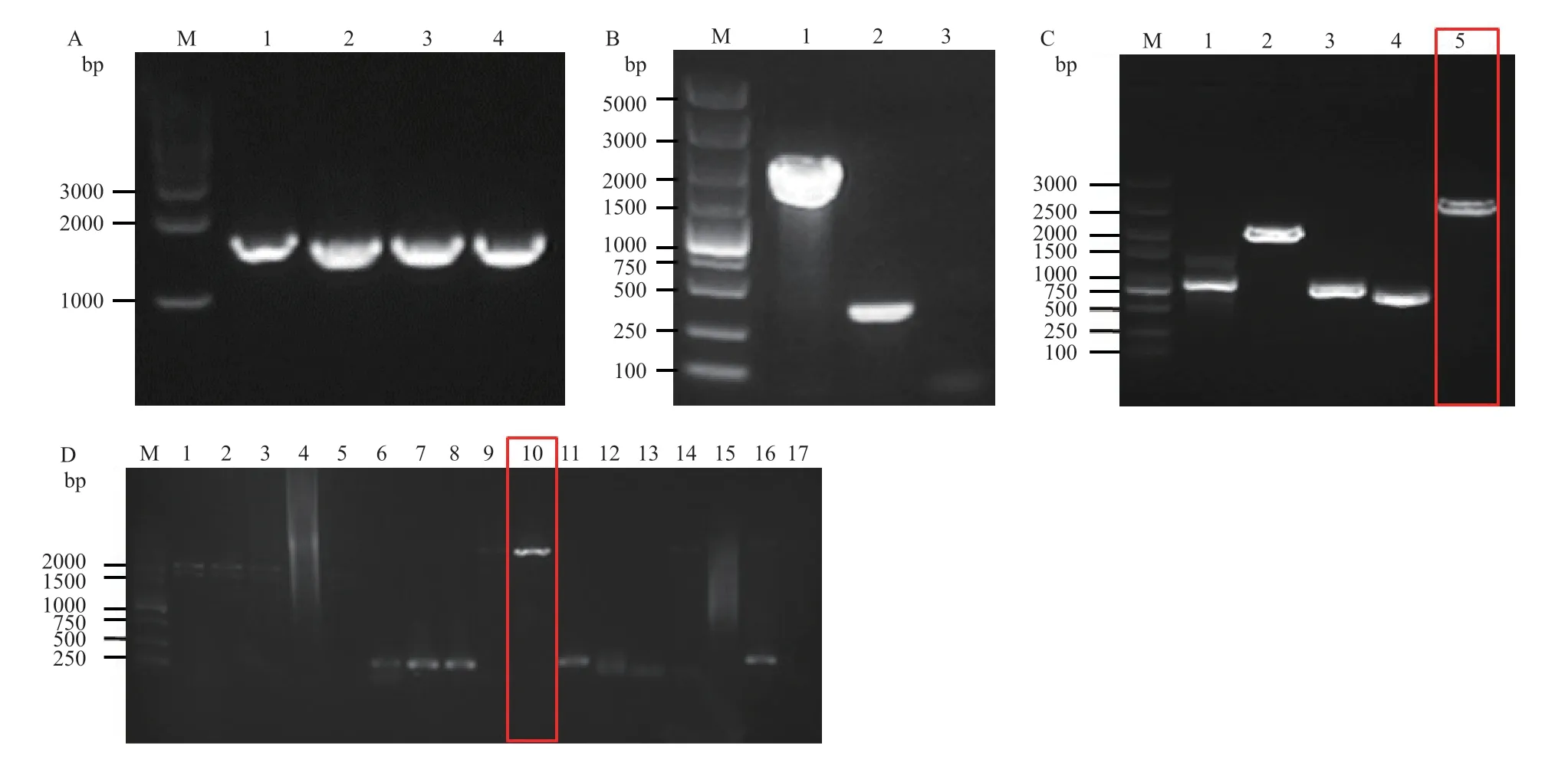
圖1 spvBC 基因編輯株的構建
2.6 鼠傷寒沙門菌質粒毒力基因spvBC回補株蛋白的誘導表達
以鼠傷寒沙門菌回補株SL1344-c-spvBC作為受試菌,敲除株SL1344-ΔspvBC做陰性對照,SL1344-c-spvB作陽性對照。經不同濃度L-阿拉伯糖誘導后Western blot 檢測SpvB 蛋白(約68 kD)表達水平。結果表明,13 mmol/L L-阿拉伯糖誘導后,回補株SL1344-c-spvBC中SpvB 蛋白正常表達(圖2-A)。以鼠傷寒沙門菌回補株SL1344-c-spvBC作為受試菌,野生株SL1344-WT 及敲除株SL1344-ΔspvBC做陰性對照。經不同濃度L-阿拉伯糖誘導后Western blot 檢測回補株SpvC 蛋白上特有的His 標簽(約30 kD)。結果表明,13 mmol/L L-阿拉伯糖可誘導回補株SL1344-c-spvBC中SpvC 蛋白正常表達(圖2-B)。以上結果表明,鼠傷寒沙門菌回補株SL1344-c-spvBC中SpvB 及SpvC 蛋白L-阿拉伯糖適宜誘導濃度為13 mmol/L。
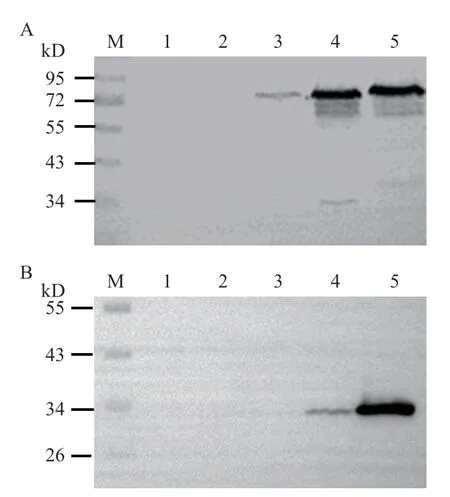
圖2 spvBC 回補株蛋白的誘導表達
3 討論
研究細菌基因功能的方法主要有基因敲除、反義RNA 干擾、轉座子插入突變、基因過表達等,但因其余方法影響因素較多、插入位點不特異和篩選方法繁瑣等缺點,基因敲除成為較可靠而常用的手段[22]。λRed 重組系統來自λ 噬菌體,克服了大腸桿菌自身核酸外切酶降解線性DNA 的缺陷,可實現幾乎任何位點的基因修飾,且在子代中穩定遺傳[23]。此外,λRed 重組系統不必依賴于酶切連接等傳統的基因工程手段,達到近乎“無痕”的敲除效果。1998 年,Murphy 等[24]首次利用Red 重組系統在大腸桿菌中進行基因敲除,由此在原核生物中建立了新的基因編輯技術。隨后廣泛應用于大腸桿菌染色體基因的敲除[18],也有文獻報道其用于沙門菌染色體基因的敲除[25],本實驗室前期利用λRed 重組系統成功敲除鼠傷寒沙門菌質粒毒力基因spvC,但未用于長約2.8 kb 的大片段基因的敲除[26]。本研究利用λRed 重組系統敲除鼠傷寒沙門菌質粒上的大片段毒力基因spvBC,為研究大片段基因的功能提供工具,也為該系統在細菌質粒基因上的應用提供思路。
影響Red 同源重組效率的因素包括線性DNA的濃度、同源片段的長度、錯配序列及內源性核酸酶等[27]。其中同源臂長度對重組效率至關重要。在大腸埃希菌中,同源臂從20 bp 增加至40 bp,同源重組效率呈指數級增長,而同源臂從40 bp 增加至1 000 bp,同源重組效率只增加10 倍。同源臂為40-50 bp 同源打靶片段即可進行高效率的重組[28]。有報道稱600-1 000 bp 的同源臂,特別適用于鼠疫耶爾森菌大片段(2-47 kb)的基因敲除[29]。本研究發現,在本實驗條件下,鼠傷寒沙門菌質粒毒力基因同源臂長度為50 bp 即可發生高效重組,既降低實驗成本,簡化實驗操作又保證了敲除效率。本研究基于λRed 重組系統構建鼠傷寒沙門菌質粒毒力基因spvBC敲除株,為研究鼠傷寒沙門菌spvBC大片段基因的致病機制提供有力的工具。
原核生物中外源基因的存在猶如細胞內多拷貝質粒一樣,對宿主是一種代謝負擔。當外源基因片段較大且表達特異蛋白時,這種代謝壓力就變得更加嚴重。研究人員將幽門螺旋桿菌HopE基因克隆到pBAD 載體上構建基因修飾菌,其編碼外膜蛋白HopE 能穩定表達并保持較高拷貝數[30]。本研究利用pBAD 原核表達載體成功構建spvBC回補株,初步摸索了該系統的實驗條件,優化回補株蛋白SpvB及SpvC L-阿拉伯糖誘導濃度。L-阿拉伯糖適宜誘導濃度為13 mmol/L,低濃度(1.3 mmol/L)L-阿拉伯糖誘導后SpvB 及SpvC 蛋白難以充分表達,而L-阿拉伯糖濃度較高(120 mmol/L)則抑制細菌生長[31]。
鼠傷寒沙門菌經糞口途徑傳播,在腸道內繁殖,粘附于腸黏膜上皮細胞,進而侵入固有層,釋放毒素,導致其充血、水腫、點狀出血等急性炎癥反應[32]。本課題組新近研究表明,鼠傷寒沙門菌質粒毒力基因spvB通過核因子E2 相關因子2(Nuclear factor erythroid-derived 2-related factor 2,NRF2)調節細胞內的鐵穩態從而影響細菌的胞內繁殖[33]。spvC可引起感染的免疫細胞逆向遷移進入血液,進而促進細菌的遠處播散,導致敗血癥[34]。已知除鼠傷寒沙門菌外,其他致病沙門菌也大都含有spv基因,以上研究提示spvB及spvC作為沙門菌保守的毒力因子,在細菌的免疫逃逸和應對宿主固有免疫應答具有重要的作用。然而spvB與spvC的關系及其在感染進程中的互作仍需進一步探究。本實驗構建鼠傷寒沙門菌質粒毒力基因spvBC敲除株和回補株,為后續深入研究spvB和spvC各自功能及其互作提供可利用的工具,也為其他原核生物的基因修飾提供科學依據和思路。
4 結論
本實驗分別利用λRed 重組系統和pBAD 原核表達載體構建鼠傷寒沙門菌spvBC基因敲除株及回補株,并發現13 mmol/L 的L-阿拉伯糖為誘導鼠傷寒沙門菌spvBC回補株中SpvB 及SpvC 蛋白正常表達的適宜濃度。鼠傷寒沙門菌spvBC基因編輯株的成功構建為原核細胞大片段基因的編輯提供了參考策略,也為后續探究spvB和spvC在沙門菌致病中的功能提供可利用的工具菌株。
