MicroRNA-182-5p對(duì)高糖誘導(dǎo)內(nèi)皮細(xì)胞損傷的影響*
2021-04-28 13:49:30崔元生尚祥李龍彪劉建雄陳陣許舒國(guó)
西部醫(yī)學(xué) 2021年4期
關(guān)鍵詞:氧化應(yīng)激糖尿病
崔元生 尚祥 李龍彪 劉建雄 陳陣 許舒國(guó)
(1.寧德師范學(xué)院附屬寧德市醫(yī)院介入科,福建 寧德 352100;2.武漢市中心醫(yī)院急診科,湖北 武漢 430014)
糖尿病是一種嚴(yán)重危害人體健康的常見(jiàn)慢性病。最新研究顯示,我國(guó)糖尿病加權(quán)患病率為11.2%,患者人數(shù)達(dá)1.56億,約占全球糖尿病總?cè)藬?shù)1/4。2017年,我國(guó)與糖尿病相關(guān)的醫(yī)療保健費(fèi)用高達(dá)1100億美元,居全球第二[1]。糖尿病血管病變是糖尿病慢性并發(fā)癥的主要表現(xiàn),其是由于慢性高血糖狀態(tài)及繼發(fā)的各種病理、生理改變而導(dǎo)致的系統(tǒng)或局部的血管損傷,是糖尿病患者致死、致殘的主要原因[2]。內(nèi)皮細(xì)胞位于血管壁內(nèi)層,長(zhǎng)期直接暴露于高血糖刺激,因此人們認(rèn)為內(nèi)皮損傷是糖尿病血管病變的始動(dòng)環(huán)節(jié)[3]。在高糖刺激下,內(nèi)皮細(xì)胞抗氧化能力降低并產(chǎn)生大量反應(yīng)活性氧(Reactive Oxygen Species,ROS),引起內(nèi)皮氧化應(yīng)激損傷和細(xì)胞死亡[4-5]。MicroRNAs是一類存在于真核生物中的非編碼單鏈小分子RNA,參與調(diào)控機(jī)體多種病理生理過(guò)程,包括血管穩(wěn)態(tài)[6-8]。MicroRNA-182-5p(miR-182-5p)在內(nèi)皮細(xì)胞中有很高的表達(dá)水平,但其在高糖誘導(dǎo)的內(nèi)皮細(xì)胞氧化應(yīng)激和死亡中作用尚不明確[9]。本研究通過(guò)建立高糖誘導(dǎo)的內(nèi)皮細(xì)胞損傷模型,旨在探討miR-182-5p對(duì)高糖誘導(dǎo)的內(nèi)皮細(xì)胞損傷的作用及可能機(jī)制。
1 材料與方法
1.1 材料 人臍靜脈內(nèi)皮細(xì)胞(Human Umbilical Vein Endothelial Cells,HUVECs)購(gòu)自美國(guó)ATCC公司;miR-182-5p的mimic、inhibitor及其對(duì)照,SIRT1小干擾RNA(Small interfering RNA against SIRT1,siSIRT1)購(gòu)自廣州銳博生物科技有限公司;CCK-8試劑盒購(gòu)自日本東仁化學(xué)科技有限公司;DMEM培養(yǎng)基(正常糖5.5 mmol/L和高糖25 mmol/L)、胎牛血清和0.25%胰蛋白酶溶液購(gòu)自美國(guó)GIBICO公司;總超氧化物歧化酶(Superoxide Dismutase,SOD)、過(guò)氧化氫酶(Catalase,CAT)、谷胱甘肽過(guò)氧化物酶(Glutathione Peroxidase,Gpx)、胱天蛋白酶-3(Caspase-3)、乳酸脫氫酶(Lactate dehydrogenase,LDH)活性檢測(cè)試劑盒和2′, 7′-二氯熒光黃雙乙酸鹽(2′, 7′-dichlorofluorescin diacetate,DCFH-DA)購(gòu)自上海碧云天公司;3-硝基酪氨酸(3-nitrotyrosine,3-NT)和丙二醛(Malondialdehyde,MDA)、SIRT1抗體、β-actin抗體購(gòu)自英國(guó)Abcam公司;逆轉(zhuǎn)錄試劑盒購(gòu)自瑞典Roche公司。
1.2 實(shí)驗(yàn)分組 HUVECs在正常糖培養(yǎng)基中培養(yǎng)48 h,經(jīng)饑餓處理12 h后隨機(jī)分為5組:對(duì)照組細(xì)胞換含有19.5 mmol/L甘露醇的正常糖培養(yǎng)基繼續(xù)培養(yǎng)24 h,高糖+mimic對(duì)照組細(xì)胞用高糖培養(yǎng)基培養(yǎng)并同時(shí)加入mimic對(duì)照試劑培養(yǎng)24 h,高糖+mimic組細(xì)胞用高糖培養(yǎng)基培養(yǎng)并同時(shí)加入mimic試劑(50 nmol/L)培養(yǎng)24 h,高糖+inhibitor對(duì)照組細(xì)胞用高糖培養(yǎng)基培養(yǎng)并同時(shí)加入inhibitor對(duì)照試劑培養(yǎng)24 h,高糖+inhibitor組細(xì)胞用高糖培養(yǎng)基培養(yǎng)并同時(shí)加入inhibitor試劑(50 nmol/L)培養(yǎng)24 h。為敲低內(nèi)皮SIRT1表達(dá),細(xì)胞先在無(wú)血清培養(yǎng)基中用siSIRT1轉(zhuǎn)染試劑(50 nmol/L)處理24 h[10-12]。
1.3 酶活性、3-NT和MDA水平檢測(cè) 細(xì)胞SOD、CAT、Gpx、Caspase-3、LDH酶活性,3-NT和MDA水平檢測(cè)均參照試劑盒說(shuō)明書(shū)進(jìn)行。
1.4 CCK-8檢測(cè) 細(xì)胞接種到96孔板并完成相應(yīng)刺激后,換無(wú)血清培養(yǎng)基并加入10 μL/孔CCK-8試劑,將細(xì)胞放回37 °C培養(yǎng)箱繼續(xù)避光孵育2 h,隨后用酶標(biāo)儀檢測(cè)450 nm處吸光度值,每組設(shè)置5個(gè)復(fù)孔[13]。
1.5 細(xì)胞內(nèi) ROS水平檢測(cè) 細(xì)胞刺激完成后換無(wú)血清培養(yǎng)基并加入5 μmol/L的DCFH-DA繼續(xù)培養(yǎng)30 min,用PBS洗滌3遍后在激發(fā)波長(zhǎng)為485 nm,發(fā)射波長(zhǎng)為525 nm的酶標(biāo)儀下檢測(cè)[14]。檢測(cè)過(guò)程中設(shè)置不含細(xì)胞的空白組,ROS水平(%)=(待測(cè)組OD值-空白組OD值)/(對(duì)照組OD值-空白組OD值)×100%。
1.6 實(shí)時(shí)熒光定量PCR 棄細(xì)胞培養(yǎng)基加入1 mL/孔TRIzol充分裂解細(xì)胞,Nanodrop 2000c檢測(cè)RNA濃度和純度,完成逆轉(zhuǎn)錄后按下列程序反應(yīng):95 ℃預(yù)變性2 min; 95 ℃×1 min,55 ℃×1 min,72 ℃×1 min,40個(gè)循環(huán);72 ℃延伸7 min。以GAPDH為內(nèi)參進(jìn)行定量檢測(cè)凋亡相關(guān)蛋白BAX和BCL-2的mRNA水平[15]。
1.7 免疫印跡 棄細(xì)胞培養(yǎng)基加入30 μL/孔裂解液,置于搖床上充分裂解20 min。用細(xì)胞刮將裂解液刮下后,轉(zhuǎn)移到新EP管內(nèi),經(jīng)超聲裂解、離心,用BCA試劑盒校正蛋白濃度,最后煮沸變性分裝保存[16]。蛋白分離采用SDS-PAGE電泳,待蛋白轉(zhuǎn)移到PVDF膜后,經(jīng)封閉、一抗和二抗孵育,用化學(xué)掃膜儀掃描并用ImageLab軟件分析定量。
1.8 熒光素酶報(bào)告基因檢測(cè) 將帶有SIRT1野生型(Wild Type,WT)和突變型(Mutant,MUT)的3′-UTR基因序列構(gòu)建到psi-CHECK2熒光素酶報(bào)告基因質(zhì)粒(美國(guó)Promega公司)中,隨后用脂質(zhì)體6000將其轉(zhuǎn)入內(nèi)皮細(xì)胞并同時(shí)予以mimic刺激,轉(zhuǎn)染48 h后收集細(xì)胞,用熒光素酶報(bào)告基因檢測(cè)試劑盒檢測(cè)熒光素酶活性[17]。

2 結(jié)果
2.1 miR-182-5p在高糖刺激的內(nèi)皮細(xì)胞中表達(dá)上調(diào) 高糖刺激可增加內(nèi)皮細(xì)胞miR-182-5p表達(dá)(P<0.05),且高糖刺激24 h的內(nèi)皮細(xì)胞miR-182-5p水平和高糖刺激48 h無(wú)差異(P>0.05),見(jiàn)圖1。
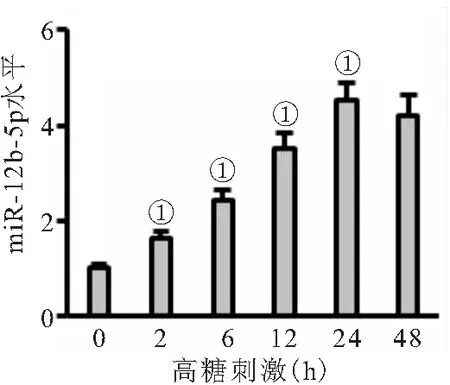
圖1 miR-182-5p在高糖刺激的內(nèi)皮細(xì)胞中表達(dá)上調(diào)
注:與對(duì)照組相比,①P<0.05
2.2 miR-182-5p mimic加重高糖誘導(dǎo)的氧化應(yīng)激,而inhibitor改善高糖誘導(dǎo)的氧化應(yīng)激 miR-182-5p mimic處理可顯著增加高糖刺激下內(nèi)皮細(xì)胞miR-128-5p表達(dá)水平,而inhibitor處理則抑制miR-128-5p表達(dá),見(jiàn)圖2。與對(duì)照組相比,高糖+mimic對(duì)照組細(xì)胞脂質(zhì)及蛋白質(zhì)氧化產(chǎn)物MDA和3-NT水平升高,抗氧化酶SOD、CAT和Gpx活性降低,ROS水平顯著增加;而使用mimic處理后則進(jìn)一步加重高糖誘導(dǎo)的內(nèi)皮細(xì)胞氧化應(yīng)激反應(yīng)(均P<0.05)。與對(duì)照組相比,高糖+inhibitor對(duì)照組細(xì)胞ROS生成也增加,抗氧化能力減弱;而使用inhibitor處理后則顯著減輕內(nèi)皮細(xì)胞氧化應(yīng)激水平(均P<0.05),見(jiàn)表1。
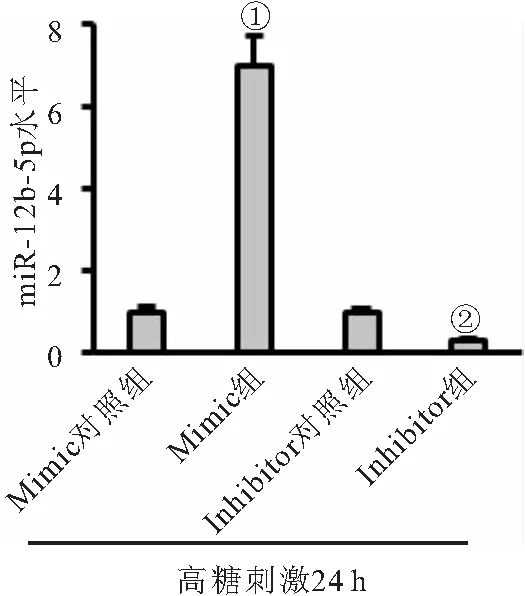
圖2 miR-182-5p的mimic和inhibitor效率檢測(cè)
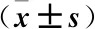
表1 各組氧化應(yīng)激指標(biāo)
2.3 miR-182-5p mimic促進(jìn)高糖誘導(dǎo)的細(xì)胞凋亡,而inhibitor減輕高糖誘導(dǎo)的細(xì)胞死亡 與對(duì)照組相比,高糖+mimic對(duì)照組細(xì)胞明顯損傷,表現(xiàn)為存活率降低、LDH釋放增加;而mimic處理則進(jìn)一步降低細(xì)胞存活率、增加LDH釋放(均P<0.05)。與對(duì)照組相比,高糖+inhibitor組細(xì)胞存活率降低、LDH釋放增加;而inhibitor處理則顯著減輕細(xì)胞損傷(均P<0.05)。高糖刺激可增加內(nèi)皮細(xì)胞Caspase-3活性,并上調(diào)促死亡蛋白BAX和抗死亡蛋白BCL-2的mRNA比值;mimic處理加重,而inhibitor處理改善高糖誘導(dǎo)的細(xì)胞凋亡(均P<0.05),見(jiàn)表2。
表2 各組細(xì)胞存活指標(biāo)
2.4 miR-182-5p通過(guò)SIRT1發(fā)揮對(duì)內(nèi)皮細(xì)胞的調(diào)控作用 在高糖刺激的內(nèi)皮細(xì)胞中,miR-128-5p mimic可減少SIRT1蛋白水平,而inhibitor處理則增加SIRT1蛋白表達(dá)(P<0.05),見(jiàn)圖3。inhibitor處理改善高糖誘導(dǎo)的內(nèi)皮細(xì)胞氧化應(yīng)激和細(xì)胞死亡;與高糖+inhibitor+siRNA組相比,高糖+inhibitor+siSIRT1組細(xì)胞ROS水平、氧化應(yīng)激產(chǎn)物MDA和3-NT生成增加,LDH釋放量也增多,而細(xì)胞存活率顯著降低,即沉默SIRT1阻斷miR-128-5p inhibitor介導(dǎo)的內(nèi)皮保護(hù)效應(yīng)(均P<0.05),見(jiàn)表3。
圖3 miR-182-5p對(duì)SIRT1蛋白表達(dá)影響
表3 各組氧化應(yīng)激和細(xì)胞存活指標(biāo)
2.5 miR-182-5p直接結(jié)合到SIRT1的3′-UTR TargetScan軟件預(yù)測(cè)發(fā)現(xiàn)miR-182-5p可直接結(jié)合到SIRT1的3′-UTR,見(jiàn)圖4A。熒光素酶報(bào)告基因檢測(cè)發(fā)現(xiàn),miR-182-5p mimic處理抑制攜帶WT報(bào)告基因的熒光素酶活性(P<0.05),而對(duì)MUT組活性沒(méi)有影響(P>0.05),見(jiàn)圖4B。
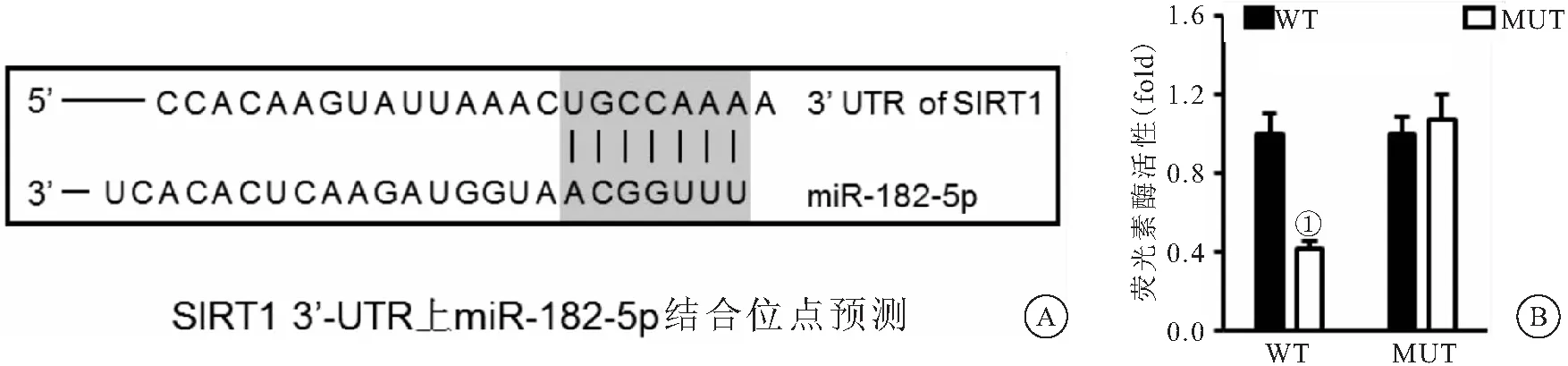
圖4 miR-182-5p直接結(jié)合到SIRT1的3′-UTR
3 討論
內(nèi)皮細(xì)胞損傷是糖尿病血管病變的關(guān)鍵病理、生理過(guò)程。在長(zhǎng)期高糖刺激下,內(nèi)皮細(xì)胞抗氧化能力減弱,ROS大量堆積從而促進(jìn)脂質(zhì)和蛋白質(zhì)等重要生物大分子發(fā)生氧化性損傷,誘發(fā)細(xì)胞死亡[2]。本研究發(fā)現(xiàn),高糖刺激可降低內(nèi)皮細(xì)胞抗氧化酶活性,細(xì)胞蛋白質(zhì)和脂質(zhì)分子氧化產(chǎn)物增加;相應(yīng)地,高糖組內(nèi)皮細(xì)胞LDH釋放量增加、細(xì)胞存活率降低且凋亡比例顯著增加。使用miR-182-5p mimic可加重高糖誘導(dǎo)的內(nèi)皮細(xì)胞氧化應(yīng)激和細(xì)胞死亡,而inhibitor處理則發(fā)揮顯著的內(nèi)皮保護(hù)效應(yīng)。此外,軟件預(yù)測(cè)結(jié)合熒光素酶實(shí)驗(yàn)證實(shí)miR-182-5p可直接結(jié)合到SIRT1的3′-UTR并抑制其蛋白表達(dá),miR-182-5p inhibitor處理則恢復(fù)內(nèi)皮細(xì)胞SIRT1水平。
SIRT1是Sirtuins家族重要成員,屬于Ⅲ類組蛋白去乙酰化酶,在細(xì)胞增殖、分化、衰老和腫瘤發(fā)生等方面均有重要調(diào)控作用[18-19]。既往研究提示,SIRT1具有顯著的抗氧化和細(xì)胞保護(hù)作用[20]。Matsui 等[21]發(fā)現(xiàn),SIRT1活化可增加抗氧化轉(zhuǎn)錄因子NRF2蛋白表達(dá)和核聚集,從而增加細(xì)胞抗氧化活性。SIRT1可促進(jìn)FoxO因子入核調(diào)控抗氧化酶SOD2轉(zhuǎn)錄[22]。此外,SIRT1可促進(jìn)凋亡相關(guān)蛋白p53去乙酰化,從而抑制其促凋亡活性;而抑制SIRT1則增加p53活性,促進(jìn)凋亡發(fā)生[23-24]。SIRT1在內(nèi)皮細(xì)胞中也有大量表達(dá),Li等[25]發(fā)現(xiàn)高糖刺激可明顯抑制內(nèi)皮細(xì)胞SIRT1表達(dá),同時(shí)伴有氧自由基大量生成和內(nèi)皮功能障礙。敲低內(nèi)皮SIRT1可增加糖尿病小鼠內(nèi)皮細(xì)胞ROS生成,加重內(nèi)皮損傷和功能障礙;而SIRT1內(nèi)皮特異性轉(zhuǎn)基因小鼠在建立糖尿病模型后則表現(xiàn)出較低的氧化應(yīng)激水平和內(nèi)皮損傷程度[26-27]。Fan等[28]近來(lái)研究發(fā)現(xiàn),激活SIRT1可減輕內(nèi)皮氧化應(yīng)激損傷,促進(jìn)內(nèi)皮存活和血管生成,從而改善糖尿病小鼠后肢缺血性損傷。本研究發(fā)現(xiàn),抑制miR-182-5p可通過(guò)激活SIRT1改善高糖誘導(dǎo)的內(nèi)皮氧化應(yīng)激和細(xì)胞死亡,而沉默SIRT1則阻斷這種內(nèi)皮保護(hù)效應(yīng)。因此,SIRT1是調(diào)控糖尿病內(nèi)皮損傷的關(guān)鍵靶點(diǎn)。
MicroRNAs是一類長(zhǎng)約19~24個(gè)核苷酸的非編碼RNA,常通過(guò)堿基互補(bǔ)配對(duì)方式與靶基因3’-UTR上種子區(qū)域完全或部分結(jié)合,在轉(zhuǎn)錄后水平調(diào)控靶基因表達(dá)[6]。近來(lái)研究提示,microRNAs參與調(diào)控細(xì)胞氧化應(yīng)激和死亡等過(guò)程。Xu等[29]研究發(fā)現(xiàn),miR-626可激活KEAP1-NRF2抗氧化信號(hào)通路減輕視網(wǎng)膜色素上皮細(xì)胞氧化性損傷。La等[30]研究提示,高糖刺激在誘導(dǎo)氧化應(yīng)激反應(yīng)的同時(shí)伴有miR-21表達(dá)上調(diào);進(jìn)一步研究證實(shí),miR-21可抑制多種抗氧化分子表達(dá),而使用miR-21抑制劑則減輕高糖誘導(dǎo)的內(nèi)皮損傷。既往有關(guān)miR-182-5p的研究大多局限在腫瘤領(lǐng)域,但越來(lái)越多研究提示,miR-182-5p在正常組織細(xì)胞的病理生理過(guò)程中也發(fā)揮重要調(diào)控作用[31]。Guzzolino等[32- 33]發(fā)現(xiàn),miR-182-5p在心臟組織中有較高表達(dá)水平,參與調(diào)控心臟發(fā)育和電活動(dòng),并可調(diào)節(jié)心肌肥厚的發(fā)生、發(fā)展。過(guò)表達(dá)miR-182-5p還可抑制腫瘤壞死因子誘導(dǎo)的氣道平滑肌細(xì)胞增殖遷移,從而參與哮喘的氣道重構(gòu)過(guò)程;miR-182-5p還是特發(fā)性肺纖維化的重要生物標(biāo)志物,抑制miR-182-5p可緩解肺纖維化進(jìn)展[34-35]。本研究發(fā)現(xiàn),miR-182-5p在高糖刺激的內(nèi)皮細(xì)胞中表達(dá)上調(diào)并抑制SIRT1蛋白表達(dá),從而加重內(nèi)皮氧化應(yīng)激損傷和細(xì)胞死亡;而抑制miR-182-5p可恢復(fù)內(nèi)皮SIRT1水平并發(fā)揮內(nèi)皮保護(hù)效應(yīng)。本研究揭示了miR-182-5p在高糖誘導(dǎo)的內(nèi)皮細(xì)胞損傷中的作用,并證明miR-182-5p是通過(guò)結(jié)合SIRT1的3′-UTR抑制其蛋白表達(dá),從而促進(jìn)高糖誘導(dǎo)的內(nèi)皮氧化應(yīng)激和細(xì)胞凋亡;為臨床治療糖尿病內(nèi)皮損傷和血管病變提供新的理論依據(jù)。
4 結(jié)論
高糖刺激增加內(nèi)皮miR-182-5p表達(dá),抑制miR-182-5p可恢復(fù)內(nèi)皮SIRT1水平,從而減輕高糖誘導(dǎo)的內(nèi)皮氧化應(yīng)激損傷和細(xì)胞凋亡;而增加miR-182-5p則降低SIRT1表達(dá),進(jìn)一步加重高糖誘導(dǎo)的內(nèi)皮損傷。靶向miR-182-5p可為糖尿病內(nèi)皮損傷和血管并發(fā)癥提供新策略。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
世界科學(xué)技術(shù)-中醫(yī)藥現(xiàn)代化(2020年2期)2020-07-25 02:05:56
西南軍醫(yī)(2016年6期)2016-01-23 02:21:19
新疆醫(yī)科大學(xué)學(xué)報(bào)(2015年10期)2015-12-26 12:33:30
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
