卷積神經網絡在核小體定位識別中的應用
2021-05-06 12:11:28崔穎施丹丹徐澤龍張兆功李建中
哈爾濱工程大學學報 2021年5期
關鍵詞:方法
崔穎, 施丹丹, 徐澤龍, 張兆功, 李建中
(1.哈爾濱醫科大學 生物信息科學與技術學院,黑龍江 哈爾濱 150086; 2.黑龍江大學 計算機科學與技術學院,黑龍江 哈爾濱 150080)
卷積神經網絡(convolutional neural networks,CNN)已被廣泛地應用于很多研究領域[1],如圖像分類、人臉識別、交通標志識別等[2]。本文將卷積神經網絡用于生物數據核小體DNA序列的識別。核小體是真核生物的染色質基本結構單元[3],由約147 bp的DNA雙鏈纏繞組蛋白八聚體約1.75圈形成,是染色體的一級結構[4]。相鄰核小體通過稱短DNA序列連接,其范圍為10 bp~100 bp。DNA序列特征一直被認為是核小體定位的重要因素。核小體參與染色質形成[5]、拮抗轉錄因子[6]、抑制基因表達[7]等重要的生物學過程,其DNA序列的精確定位不僅影響基因表達調控[8],對DNA復制[9]、DNA修復[10]及重組[11]等也有重要作用。當前國內外核小體定位研究大多針對開發分類算法,例如通過信息熵[12]、堿基對偏轉角度[13]等方法來表示核小體DNA序列特征[14],進而訓練分類器。本文基于Z曲線理論將核小體DNA序列轉換為三維空間曲線坐標,應用卷積神經網絡模型訓練和檢驗,獲得了較好的分類效能,為今后的核小體定位研究提供一些參考。
1 基于Z曲線理論的卷積神經網絡方法(ZCN)
1.1 Z曲線理論及轉換公式
Z曲線理論基于正面體表示堿基對稱性[15],將DNA 序列用4 種字母符號表示為一維序列,利用這種形式來表示任意長度的DNA 序列,能夠顯示出DNA序列的新特征,且根據正四面體的對稱性可以證明,每條序列對應唯一一條Z曲線。對于一條長為N的單鏈DNA序列,它的三維坐標可表示方法為:
(1)
每次從第1個堿基到第n個堿基,分別統計1~n這個子序列中4種堿基積累出現的次數,記為An、Cn、Gn、Tn。Xn、Yn、Zn的取值范圍為[-n,n],對其進行標準化處理, 將Xn、Yn、Zn的值都除以n得到xn、yn、zn,使其范圍處于[-1,1]:
(2)
1.2 卷積神經網絡分類器
卷積神經網絡是一種前饋神經網絡[16],包括卷積層(convolutional layer)和池化層(pooling layer),布局更接近于真實生物神經網絡,能降低特征提取和分類過程中數據的復雜程度[17]。卷積神經網絡的構建包括創建卷積層、創建線性整流層、創建池化層以及創建全連接層4個步驟。
本文基于Z曲線理論,應用卷積神經網絡提出一種新的核小體定位識別方法,簡稱為ZCN,該方法的流程圖如圖1所示。分類器的構建過程使用R軟件包“mxnet”進行訓練和驗證,采用十倍交叉驗證方法進行效果評估,取10次驗證的平均結果為一次最后結果,同時,為了減少由于隨機分類而帶來的結果誤差,隨機重復進行50次十倍交叉驗證。具體過程如下:將Z曲線模型投入卷積層構建模型,卷積核大小為3,并選定卷積核個數為300創建卷積層;然后加入非線性函數即雙曲正切函數創建線性整流層;再采用最大池化,步長設置為1創建池化層;最后,每一個結點都與上一層的所有結點相連,用來把前邊提取到的特征綜合起來,最終得到全連接層,并用“softmax”函數構建分類器模型,使用驗證集數據進行分類器檢驗;最后,通過驗證數據集進行驗證。
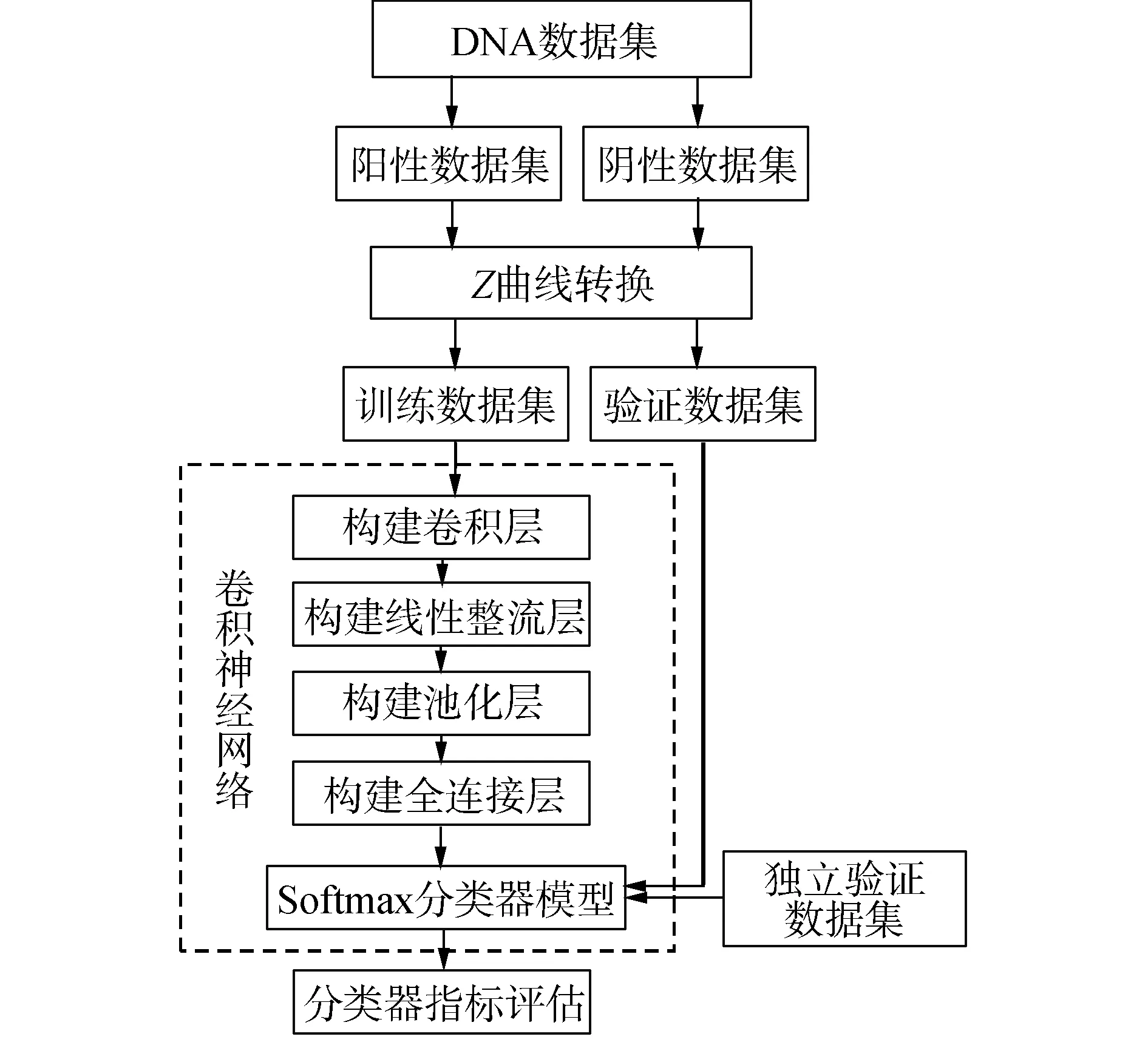
圖1 基于Z曲線的卷積神經網絡流程Fig.1 Flow chart of convolutional neural network based on Z-curve
1.3 分類器模型性能評估
為評價ZCN方法的分類效果,使用敏感性(sensitivity, Sn)、特異性(specificity, Sp)、準確率(accuracy,Acc)和Matthews相關系數(matthews correlation coefficient,MCC)及ROC(receiver operating characteristic)曲線面積AUC(area under ROC)來作為評價參數[18],前3個指標通常被用于在統計預測理論中從不同角度衡量預測系統性能為:
(3)
式中:TP表示真陽性(true positive, TP)數量;FP表示假陽性(false positive, FP)數量;TN表示真陰性 (true negative, TN) 數量;FN表示假陰性(false negative, FN)數量。
2 實驗結果
2.1 數據來源及預處理
2.1.1 酵母數據集
從文獻[19]中獲得酵母(Saccharomyces cerevisiae,S.cerevisiae)核小體數據,含有5 000條核小體DNA序列作為陽性數據集,與5 000條連接DNA序列作為陰性集,每條序列長為150 bp,記為數據集S1;同時,采用文獻[20]中的1 880條核小體DNA序列集和1 740條連接區DNA序列集,記為數據集S2。
2.1.2 人類、線蟲和果蠅數據集
從Guo文獻中獲得人類(H.sapiens)、線蟲(C.elegans)和果蠅(D.melanogaster)的數據集[21],人類共有2 273條核小體DNA序列集和2 300條連接區DNA序列集,線蟲共有2 067條核小體DNA序列集和2 108條連接區DNA序列集,果蠅共有2 400條核小體DNA序列集和2 350條連接區DNA序列集,用于檢驗ZCN方法分類效能和可推廣性。
2.1.3 酵母全基因組數據
從UCSC數據庫獲取釀酒酵母全基因組序列數據[22],包含17條染色體序列,其網址為:http://hgdownload.soe.ucsc.edu/downloads.html(版本:SacCer_Apr2011 sacCer3),使用其中16條染色體數據;另外從SGD(Saccharomyces Genome Database)數據庫獲得酵母基因GAL1和基因GAL10的DNA序列數據,用于核小體預測,其網址為http://www.yeastgenome.org/。
2.1.4 酵母全基因組核小體數據集
從Xu Zhou與Erin O′Shea的文獻中獲得61 532條酵母核小體位置信息數據[23],這是一套全基因組的核小體數據集,用于檢驗預測結果。
2.2 分類器識別結果與分析
2.2.1 ZCN方法在酵母數據集中的實驗結果
酵母數據集S1的核小體DNA序列和連接區DNA序列經過卷積神經網絡進行訓練和驗證,結果如圖2(a),敏感性Sn、特異性Sp、準確率Acc和MCC值分別為0.91、0.88、0.90、0.80,ROC曲線下面積AUC值為0.96,面積最高值達到0.970 4,如圖2(b),這表明ZCN方法在酵母的核小體定位識別中的效能良好,能夠較好地識別出核小體序列與連接區序列。
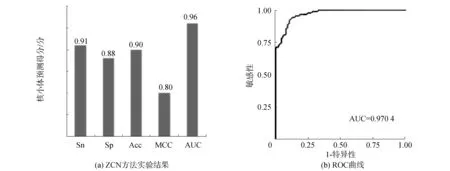
圖2 ZCN方法實驗結果和ROC曲線(S1)Fig.2 Results of ZCN method and receiver operating characteristic(S1)
酵母數據集S2的核小體DNA序列和連接區DNA序列,分別經過Z曲線轉換得到三維空間坐標矩陣數據集,投入訓練出的分類器中進行分類,通過十倍交叉驗證,結果如圖3(a)所示,Sn、Sp、Acc和MCC分別達到0.97、0.84、0.90、0.82。ROC曲線面積AUC值最高達到0.972 3,如圖3(b)所示,表明ZCN方法在酵母核小體定位中再次取得較好識別效果,且各項性能指標穩定。實驗表明ZCN方法在酵母中具有較好的應用效果。
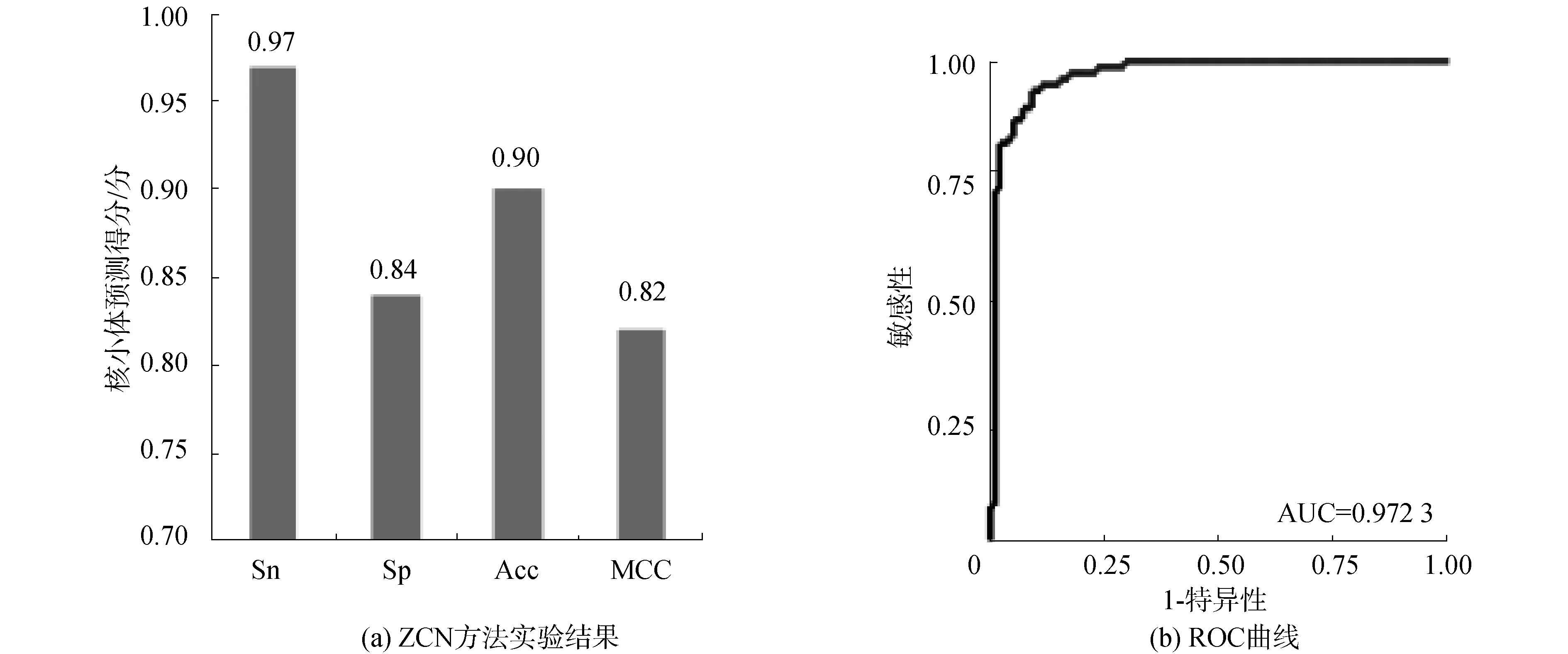
圖3 ZCN方法實驗結果和ROC曲線(S2)Fig.3 Results of ZCN method and receiver operating characteristic(S2)
2.2.2 ZCN方法在人類、線蟲和果蠅中的實驗結果
應用ZCN方法識別人類、線蟲和果蠅3個物種的核小體定位,得到人類、線蟲和果蠅的ROC曲線下面積AUC值分別為0.796、0.940和0.772,如圖4所示,Sn、Sp和Acc值如表1所示。ZCN方法不僅可應用于酵母也可應用于其他多個物種的核小體定位識別,分類效能良好且具有可靠的推廣性。
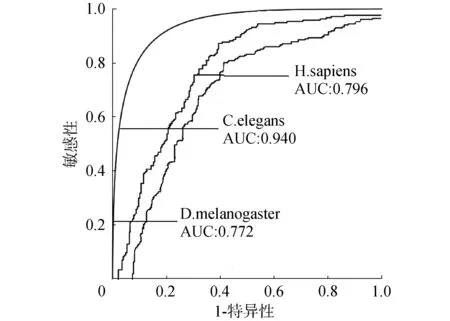
圖4 人類、線蟲和果蠅的ROC曲線面積Fig.4 ROC curve areas for H.sapiens, C.elegans and D.melanogaster
2.2.3 ZCN方法與其他識別方法的比較
將ZCN方法與其他方法進行結果比較,包括iNuc-STNC方法[21]、iNuc-PseKNC方法[18]、3LS方法和LeNup[14],這4種方法沒有酵母實驗結果,因此只進行人類、線蟲和果蠅的核小體定位識別結果比較;另外,LeNup方法采用20倍交叉驗證,而3LS、iNuc-PseKNC和iNuc-STNC方法采用Jackknife檢驗方法,ZCN方法采用10倍交叉驗證方法,其訓練數據集在樣本大小和訓練次數上均小于上述4種方法,如表1所示。
表1ZCN方法與其他方法的實驗結果比較
Table1ComparisonofexperimentalresultsbetweenZCNmethodandothermethods
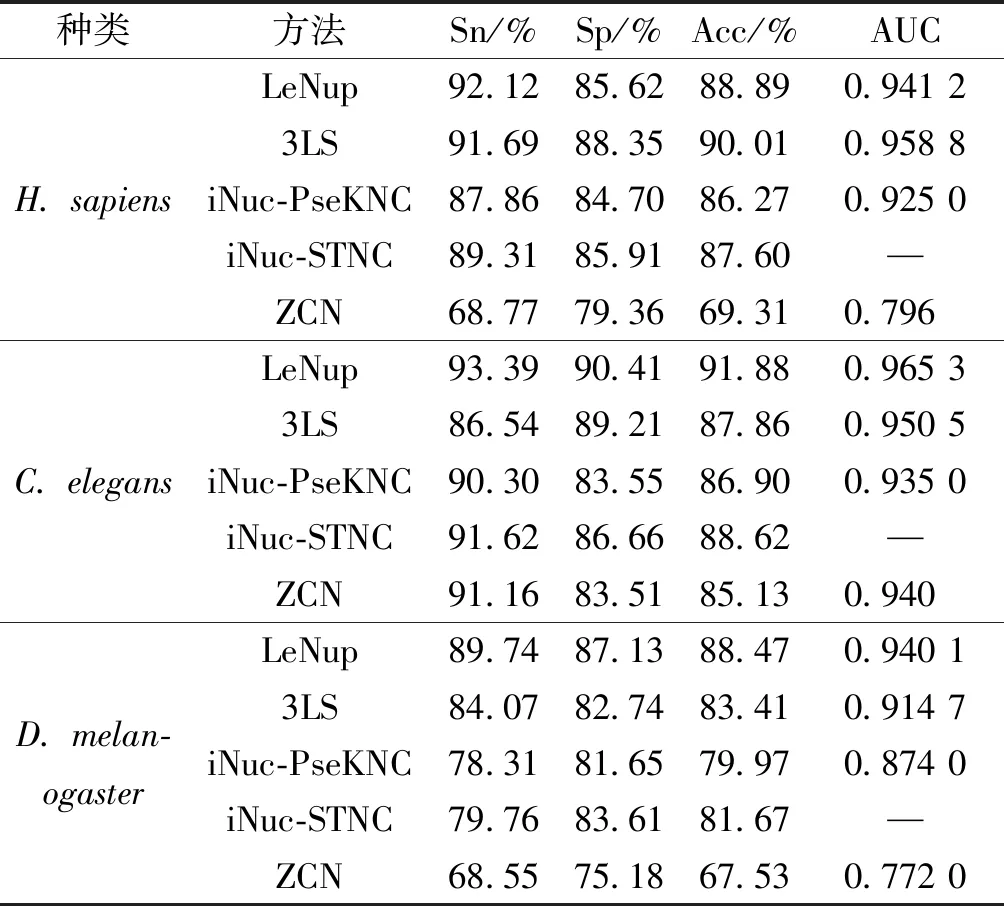
種類方法Sn/%Sp/%Acc/%AUCH. sapiensC. elegansD. melan-ogasterLeNup92.1285.6288.890.941 23LS91.6988.3590.010.958 8iNuc-PseKNC87.8684.7086.270.925 0iNuc-STNC89.3185.9187.60—ZCN68.7779.3669.310.796LeNup93.3990.4191.880.965 33LS86.5489.2187.860.950 5iNuc-PseKNC90.3083.5586.900.935 0iNuc-STNC91.6286.6688.62—ZCN91.1683.5185.130.940LeNup89.7487.1388.470.940 13LS84.0782.7483.410.914 7iNuc-PseKNC78.3181.6579.970.874 0iNuc-STNC79.7683.6181.67—ZCN68.5575.1867.530.772 0
ZCN方法在人類和果蠅數據集中,敏感性Sn、特異性Sp、準確性Acc 3項指標略低,ROC曲線面積AUC值分別達到0.796和0.772,而iNuc-STNC方法沒有給出AUC值;在線蟲數據中,ZCN方法的敏感性Sn高于3LS方法,特異性Sp、準確性Acc與iNuc-PseKNC方法基本一致,ROC曲線面積AUC值0.940略高于iNuc-PseKNC方法。ZCN方法在訓練數據集大小和訓練次數均小于其他4種方法,但在單項指標上表現較好,且各項指標穩定,特別地在酵母中取得較好實驗結果,表明ZCN方法有識別較好效能和可推廣性。
2.3 酵母核小體預測結果與分析
2.3.1 核小體序列預測候選序列集
通過滑窗法,設置滑動窗口大小為150 bp,步長為1 bp,按染色體提取出核小體DNA序列候選預測集,如表2所示,共得12 068 942條DNA序列作為候選預測集,將每條DNA進行Z曲線轉換得到坐標矩陣,投入到ZCN方法訓練的模型中進行預測。
表2酵母全基因組核小體定位候選預測集
Table2CandidatepredictionsetofnucleosomelocalizationinthewholegenomeofS.cerevisiae
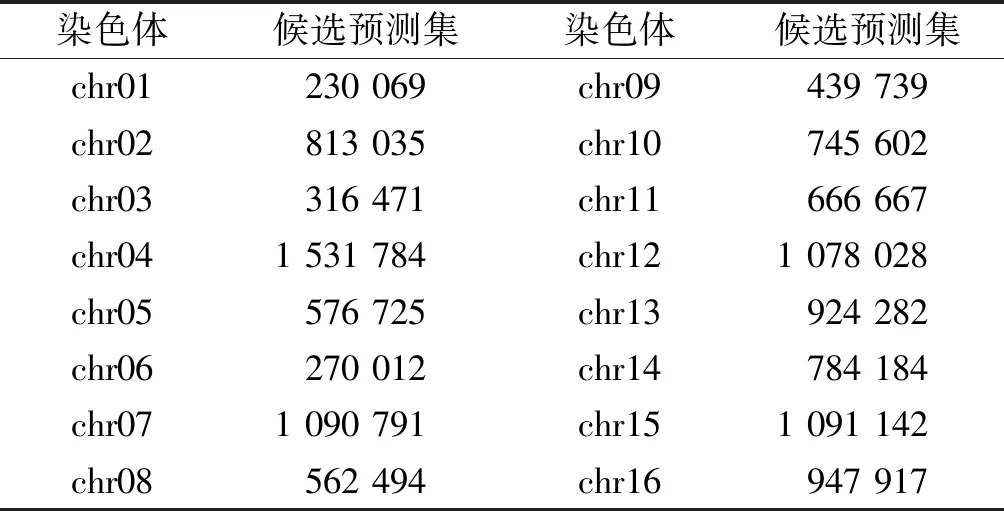
染色體候選預測集染色體候選預測集chr01230 069chr09439 739chr02813 035chr10745 602chr03316 471chr11666 667chr041 531 784chr121 078 028chr05576 725chr13924 282chr06270 012chr14784 184chr071 090 791chr151 091 142chr08562 494chr16947 917
2.3.2 核小體預測去冗余篩選方法
滑窗法獲得候選序列集存在大量相似序列和冗余數據,為減少預測結果中的重復和冗余,提出基于染色體上每個堿基位置的去冗余策略,對序列陽性集進行篩選,保留預測為核小體的DNA片段,將這些片段回拼至全基因組對應位置上。
篩選方法思想如下:1)每條染色體上的每條候選預測序列,除去首尾2條候選預測序列的75 bp堿基外,每條候選預測序列的每個堿基,將其擴展前后共150 bp序列提取出來投入ZCN訓練模型中進行預測。若預測為核小體,則將這一堿基標記為核小體。因此,除了每條染色體序列最開始的149個堿基與末尾149個堿基,相當于在考慮周圍序列信息的條件下,對染色體上單個堿基進行150次記分。堿基所對應的得分越高,該堿基前后共150 bp堿基所對應的150條序列被分類為核小體序列的數量越多,即這個位置的堿基更傾向于落到核小體序列片段;2)如果一條DNA片段被分類為核小體,則該序列對應堿基150 bp范圍內的所有堿基得分加1,否則加0,在對所有序列進行分類之后,所有位置的堿基的得分范圍為0~150,設定初始閾值75,當每個位置的堿基得分閾值大于或等于75時,認為該位置堿基更加傾向于落在核小體區域,逐漸提高閾值,找到可以降低假陽性的更加嚴格的閾值,最后統計篩選出得到去除有重復的相鄰候選序列,得到預測的核小體數量,如圖5所示,經過28次計算,最后選擇閾值為93篩選所得9 229 869個堿基位置作為核小體預測結果。
2.3.3 核小體預測結果驗證
為檢驗其與驗證集在單條染色體上的核小體結果的一致性,使用一套獨立數據集(61 532條核小體序列)來驗證結果,繪制酵母單條染色體上預測堿基與實際堿基位置統計圖。如圖6所示,發現不僅每一條染色體上的2個數據量大小相近,全基因組上的整體數據趨勢也相同,如圖6(a)所示;為了精確觀察具體差異程度,計算出每條染色體2類數據的差值在真實堿基數量上的比例,結果顯示差值比例都小于4%,最大值為3.7%,最小值僅為0.1%,如圖6(b)所示,預測結果與驗證數據有較好的一致性,表明核小體去冗余篩選方法得到的閾值具有可靠性。
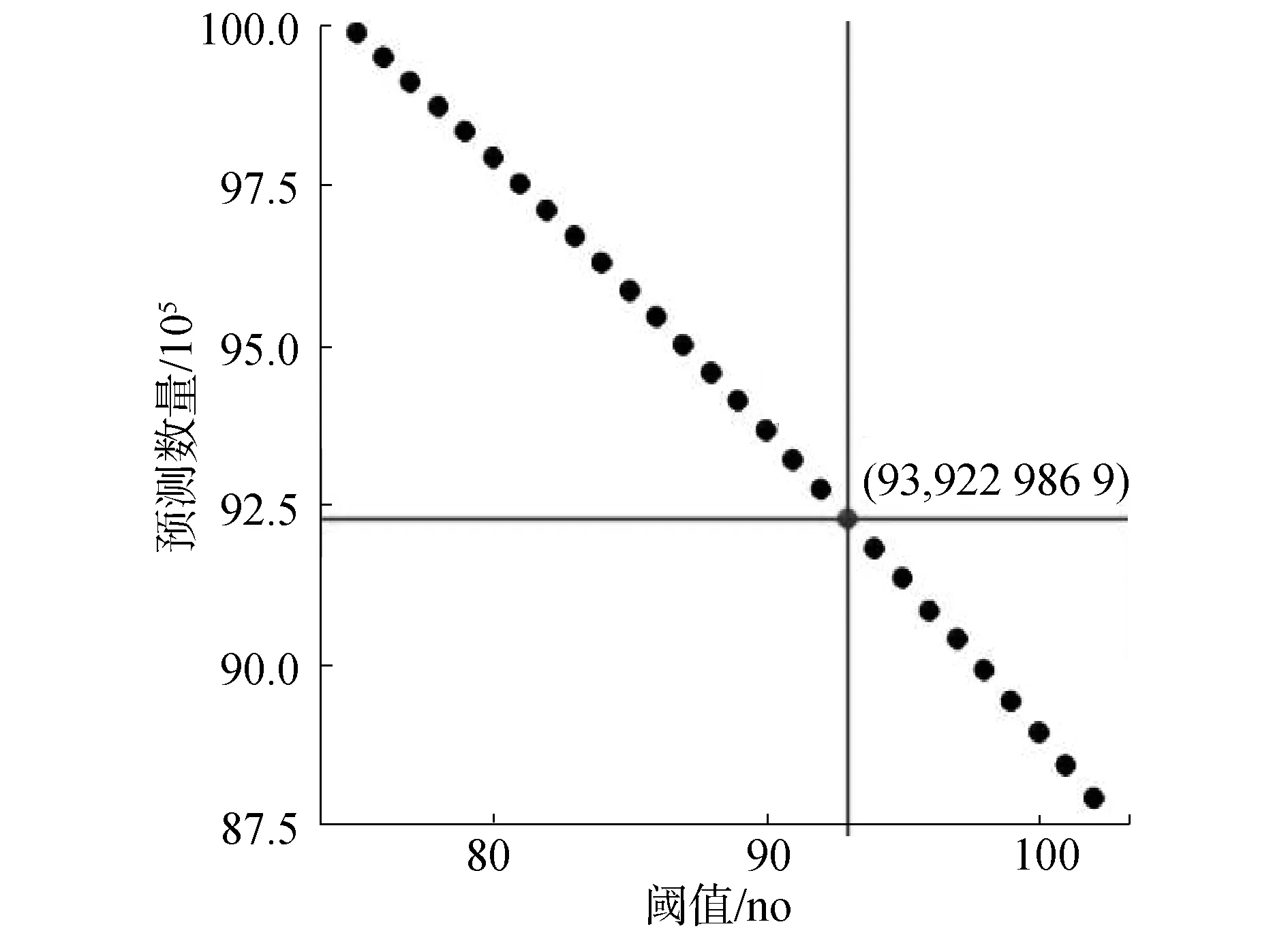
圖5 候選預測結果閾值篩選Fig.5 Threshold selection of candidate prediction result

圖6 預測位置與實際位置的驗證統計Fig.6 Statistics of predicted position and actual position
計算單條染色體預測位置與實際位置的交集,以檢驗核小體定位的準確性,可見位置的重疊程度較高,如表3所示。
表3酵母核小體定位的全基因組位點驗證
Table3Wholegenomesiteverificationofyeastnucleosomepositioning

染色體預測結果集驗證集交集準確率/%chr01174 619174 300136 46078.29chr02633 933621 000500 11580.53chr03232 690241 650185 05676.58chr041 148 1951 173 300910 12377.57chr05435 367443 850345 53977.85chr06207 727207 900166 16979.93chr07825 042828 000645 70877.98chr08433 723428 550343 36180.12chr09344 939336 600271 97780.80chr10581 059571 200451 14978.98chr11504 252507 300388 38276.56chr12826 454818 700647 84279.13chr13705 540711 450561 77178.96chr14619 530598 650487 81481.49chr15835 707838 650661 40878.87chr16721 092728 700572 72778.60總數9 229 8699 229 8007 275 60178.83
單條染色體交疊堿基數量真實驗證數據集堿基數的比例最低為76.56%,最高81.49%,平均值為78.83%,根據比例值,對預測結果集與驗證集做Wilcoxon秩和檢驗,P-value為 0.690 5,差異不顯著;根據位置,對預測結果集數據與驗證集的核小體起始和終止位置,計算其皮爾森相關系數(Pearson correlation coefficient)以驗證預測結果與驗證結果的相似性,發現每條染色體上的皮爾森相關系數值均大于0.99,P-value<2.2×10-16,這充分說明預測結果集與驗證集的相關性接近于100%。根據結果分析,發現核小體序列能夠很大范圍地被預測出來,通過閾值篩選后,其驗證結果較好,說明ZCN方法可以進行全基因組核小體定位預測,并且在訓練集大小為5 000條核小體序列和5 000條連接序列的情況,不僅完成全基因組6萬多條核小體序列預測,并取得78.83%的預測準確率,結果證實ZCN方法預測效果較好。
2.3.4 酵母基因GAL1與基因GAL10的預測結果
從SGD數據庫獲得基因GAL1和GAL10的DNA序列數據及位置數據,用ZCN方法在這兩個基因上進行核小體定位預測和驗證,預測過程與全基因組上核小體預測過程一致,GAL1原基因長度為1 587 bp(chrII:279021-280607),加上下游1 kb,共為3 587 bp(chrII:278021-281607),GAL10原基因長度為2 100 bp(chrII:276253-278352),加上下游1 kb,共為4 100 bp(chrII: 275253-279352)。
預測結果如圖7所示,計算該基因上預測位置與實際位置的交集,及交集在預測集與真實集中的占比。

圖7 預測位置與實際位置的驗證統計Fig.7 Statistics of predicted position and actual position
圖7中,粗線表示預測核小體堿基中交集位點的含量,即預測核小體堿基的準確率;細線表示在真實核小體堿基中交集位點的比例,也可以表示每個閾值下核小體的檢出率,它越來越低是因為隨著閾值升高,預測堿基位點數量逐漸降低,導致交集數量減少,但作為分母的真實核小體堿基數量一直不變。當閾值設為1時,表示以卷積神經網絡模型一次分類即為預測結果,此時在基因GAL10序列中有3 374個堿基被預測為核小體位點,其中有2954(約87.55%)個為真實的核小體堿基位點,占真實的核小體堿基位點數據集(3 241個)的91.14%。而在基因GAL1序列中有2 699個堿基被預測為核小體位點,其中有2 213(約81.99%)個為真實的核小體堿基位點,占真實的核小體堿基位點數據集(2 700個)的81.96%。可見位置的重疊程度較高,說明在不考慮假陽性的情況下,核小體序列能夠很大范圍地被預測出來。
為了降低假陽性,逐漸提升閾值來綜合考慮多次分類的結果,雖然核小體的預測位點數量與堿基交集數量逐漸減少,但該閾值下預測集中的準確性卻逐漸增高至92.53%,甚至100%(GAL10中:準確性最低為87.25%,最高為92.53%,平均值為89.16%;GAL1中:準確性最低為81.38%,最高為100.00%,平均值為86.25%),結果證實ZCN方法預測效果較好。盡管真實的核小體位點數量的檢出率顯著降低,但是預測集中的準確性卻波動相對較小,這意味著預測結果中假陽性比例不大,且今后也許可以根據核小體所需數量來確定閾值,即使將閾值設置為個位數,預測的準確性都能高于81%。根據預測核小體堿基位點的得分,繪制峰值圖譜,如圖8所示,結果顯示在基因GAL1周圍上下游出現7個峰值,基因GAL10周圍上下游出現11個峰值,即預測的核小體定位數量,這與理論分析基本一致,再次說明ZCN方法的預測效能較好。
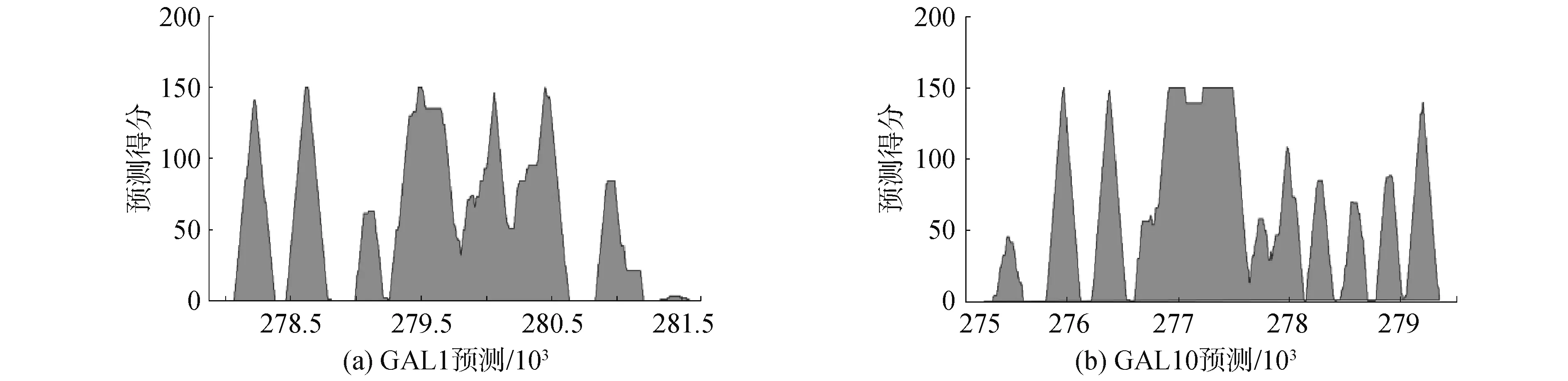
圖8 基因GAL1和GAL10上的核小體定預測圖譜Fig.8 Predicted map of nucleosome positioning on GAL1 and GAL10 genes
3 結論
1)將ZCN方法用于酵母核小體定位識別,通過2套數據實驗,結果顯示ZCN方法在酵母中取得較好的識別效果。
2)將ZCN方法能夠推廣到其他物種,包括人類、線蟲和果蠅的核小體定位識別中,與其他4種方法進行性能比較,結果顯示ZCN方法能夠很好進行物種推廣。
3)將ZCN方法用于酵母全基因組核小體定位預測,又提出一個基于堿基位置的閾值篩選方法,既獲得高質量的核小體偏好堿基,又降低數據冗余性,將篩選后的預測結果與實驗獲得的核小體數據(驗證集)比較驗證,同時通過預測基因GAL1和GAL10周圍的核小體位點,進而獲得核小體定位的位置分布情況,均顯示出預測結果集與驗證集的具有較好的一致性和較高的準確,說明ZCN方法能夠很好地完成核小體定位預測。
ZCN方法獲得較好的實驗結果,是由于Z曲線的三維坐標矩陣很好地展現出DNA序列特征,卷積神經網絡很好地完成了這些特征的訓練,因此,實驗結果顯示各項性能指標都取得不錯效果。ZCN方法對核小體定位預測和核小體功能研究具有重要的參考和指導意義,特別地,對于深入理解基因表達的后續步驟以揭示控制核小體定位所涉及的機制也有重要作用。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
