結核分枝桿菌甲硫氨酰tRNA合成酶與底物及其類似物的結合比較
2021-05-06 08:59:56任偉宏陳明心
中國人獸共患病學報 2021年4期
王 煒,任偉宏,陳明心,蔣 露,張 岱,陳 磊
氨酰tRNA合成酶(aminoacyl-tRNA synthetases, aaRS)是一類細胞內高度保守蛋白酶家族,參與到生命遺傳信息翻譯過程中,是確保細胞遺傳信息正確翻譯的酶。不同物種來源的MetRS具有結構上的多樣性,適合特異性抑制劑的篩選,已被證明是理想的抗生素作用靶點[1]。在近幾年的aaRS抑制劑研究中,發現的一些氨酰tRNA合成酶抑制劑,具有高選擇性和強抑制能力。AN3016是一種亮氨酰tRNA合成酶抑制劑,動物實驗顯示,其抑制結核分枝桿菌的能力強于異煙肼[2]。
結核分枝桿菌MetRS(MycobacteriumtuberculosisMethionyl-tRNA synthetase, MtMetRS)的研究始于上個世紀,相關文獻報道其氨酰化反應后就沒有新的研究報道。MtMetRS研究進展緩慢的原因與其重組蛋白制備困難有關[3]。由于MetRS抑制劑存在專一性強、抗菌譜覆蓋面窄的問題。為了篩選結核分枝桿菌MetRS的抑制劑,首先需要大量獲得MtMetRS。我們通過構建不同重組質粒,進行重組蛋白表達篩選,最終解決了MtMetRS重組蛋白制備問題,為抑制劑的篩選提供基礎條件。此外,我們使用Thermal Shift Assay(TSA)驗證了重組MtMetRS與底物及其類似物的結合能力,為抑制劑的選擇提供方向。
1 材料與方法
1.1材料 pET32a、pQE60、pGEX-6P-1、pTrcHisB質粒、M15大腸桿菌、BL21大腸桿菌本實驗室保存。高保真DNA聚合酶、限制性核酸內切酶、T4 DNA連接酶、DNA marker購自Takara公司。質粒提取、凝膠回收試劑盒購自Axygen公司。引物及基因合成、測序在生工生物工程(上海)股份有限公司完成。青霉素、IPTG購自索萊寶公司。Milipure超濾管購自默克公司。HisPur Ni-NTA樹脂、蛋白質Marker購自Thermo Scientific。ATP、Met、ADO、AMP-PNP、Sypro Orange購自Merck公司。
1.2 方 法
1.2.1MetRS序列分析 氨基酸序列決定蛋白質結構。為了分析人MetRS和結核分枝桿菌MetRS的差異,選用GenBank登錄號為AAH15011.1、AAH09115.1、WP_003911321.1氨基酸序列為分析對象。氨基酸序列以FASTA格式整理為一個文件。使用MEGA軟件中的MUSCLE[4]Neighbor-Joining算法對MetRS氨基酸序列進行多序列比對分析。以比對好的文件和人胞質MetRS晶體結構(PDB ID:5GL7)為材料,使用ESPript網絡服務制作基于結構的多序列比對圖,并指示出參與底物結合的氨基酸殘基。
1.2.2重組基因的表達篩選 根據GenBank登錄號CP003248.2核酸序列,由公司合成MtMetRS基因序列并插入到pET32a的NcoI、XhoI限制性核酸內切酶識別序列之間。通過PCR引物設計,在MtMetRS基因的兩端加入相應的限制性核酸內切酶識別序列,通過限制性核酸內切酶酶切及T4連接酶鏈接,MtMetRS基因連接到pQE60、pGEX-6P-1、pTrcHisB載體上。使用菌落PCR鑒定陽性克隆后送公司測序確認MtMetRS基因開放讀碼框是否正確。pQE60-MtMetRS重組質粒的啟動子為T5啟動子,使用M15宿主菌進行誘導表達。pET32a-MtMetRS的T7啟動子、pGEX-6P-1-MtMetRS的tac啟動子、pTrcHisB-MtMetRS的trc啟動子可以在BL21宿主菌中轉錄表達。pQE60-MtMetRS/M15、pET32a-MtMetRS/BL21、pGEX-6P-1-MtMetRS/BL21、pTrcHisB-MtMetRS/BL21,在18 ℃、250 r/min、1 mM IPTG條件下過夜誘導重組MtMetRS表達。誘導后4 ℃離心收集細菌,菌泥重懸到500 mmol/L NaCl、50 mmol/L Tris pH8.0裂解緩沖液中。菌液超聲裂解后,15 000 g離心45 min去除未破碎的細菌及不溶性顆粒,可溶性部分用Ni-NTA樹脂結合。首先用20 mL裂解緩沖液洗滌樹脂,然后用15 mL含25 mmol/L咪唑的相同緩沖液洗滌。最后,用10 mL含300 mmol/L咪唑的緩沖液洗脫樹脂結合的蛋白。分別取裂解液和Ni-NTA樹脂洗脫液加入4×蛋白上樣緩沖液,90 ℃加熱10 min后用SDS-PAGE分析。
1.2.3重組MtMetRS的制備 通過表達篩選,確定使用pQE60-MtMetRS質粒制備重組MtMetRS。使用上面相同的條件誘導表達2 L菌液,經超聲破碎、高速離心、Ni-NTA樹脂結合與洗脫后,使用截留分子量為50 kDa的Milipure超濾管濃縮洗脫液至1.5 mL。濃縮液注入kta層析儀中,使用100 mmol/L NaCl、50 mmol/L Tris pH8.0緩沖液進行色譜分離。收集洗脫峰樣品,使用Milipure超濾管濃縮。濃縮蛋白經Nanodrop調整濃度為10 mg/mL。50 μL分裝后經液氮速凍后-80 ℃保存。
1.2.4MtMetRS與配體的結合分析 使用100 mmol/L NaCl、50 mmol/L Tris pH8.0緩沖液稀釋Sypro Orange到10×。重組MtMetRS蛋白稀釋到20 μmol/L濃度后與5 mmol/L濃度的ATP、Met、AMP-PNP、ADO、Met+ATP、Met+ADO、Met+AMP-PNP按照1∶1體積比混合,在冰上孵育15 min,然后與10× Sypro Orange按照1∶1體積比混合后冰上孵育15 min。在Bio-Rad CFX實時定量PCR儀上設置程序:溫度范圍20~80 ℃,從20 ℃起始,溫度每升高0.5 ℃,撫育1 min,使用Cal Gold540通道檢測Sypro Orange熒光信號。
2 結 果
2.1多序列比對結果 MetRS的氨基酸多序列比對結果如圖1所示,紅色五星指示出參與結合中間產物MetAde的氨基酸殘基。MetRS雖然催化相同的化學反應,但這些氨基酸殘基在人MetRS和結核分枝桿菌MetRS中并不完全一致。這個差異提示不同MetRS結合相同化合物的能力可能存在差異。而這種差異是設計篩選特異性、低毒性MetRS抑制劑的前提。
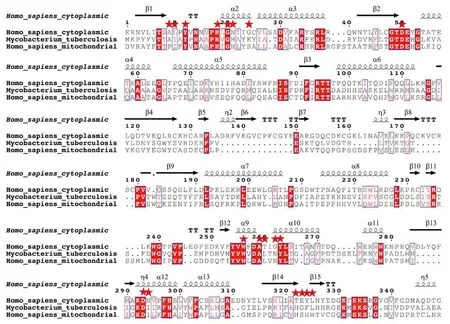
圖1 MetRS的多序列比對Fig.1 Multiple sequence alignment of MetRS
2.2重組基因載體的構建 針對pQE60、pGEX-6P-1、pTrcHisB載體多克隆位點的限制性核酸內切酶識別序列設計MtMetRS基因引物。以pET32a-MtMetRS質粒為模板擴增MtMetRS基因。擴增結果如圖2所示,在1 500 bp marker旁有一條帶,與MtMetRS基因大小近似。PCR產物經瓊脂糖凝膠純化后,經限制性核酸內切酶消化后連接入相應載體中。重組質粒經測序驗證讀碼框完全正確。
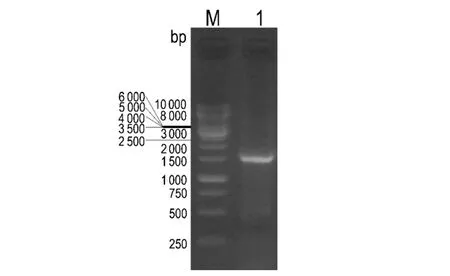
M: The product of MtMetRS gene amplification;1: GeneRuler 1 kb Marker圖2 MtMetRS基因 PCR擴增結果Fig.2 Result of MtMetRS gene amplification
2.3重組載體的表達篩選 為獲得可溶性重組蛋白,在重組質粒表達篩選時我們只驗證細菌裂解液上清是否可以純化到重組蛋白。重組質粒轉化宿主菌后誘導表達500 mL菌液,經超聲裂解后使用Ni-NTA樹脂純化。分別取細菌裂解液和Ni-NTA樹脂洗脫液,使用SDS-PAGE分析。電泳結果如圖3所示,第3、5泳道分別是pQE60-MtMetRS、pTrcHisB-MtMetRS質粒表達純化的重組蛋白,與55 kDa蛋白marker位于同一水平。1、7泳道分別是pET32a-MtMetRS、pGEX-6P-1-MtMetRS質粒表達純化的重組蛋白。2、4、6、8泳道分別為pET32a-MtMetRS/BL21、pQE60-MtMetRS/M15、pTrcHisB-MtMetRS/BL21、pGEX-6P-1-MtMetRS/BL21誘導表達后的裂解液。由于pTrcHisB-MtMetRS質粒表達的重組蛋白氨基端含有一個短的標簽。為避免重組蛋白氨基端多余多肽對蛋白質活性的影響,我們最終選擇pQE60-MtMetRS質粒作為重組蛋白MtMetRS的生產質粒。

M:Protein marker;1:Recombinant MtMetRS expressed by pET32a-MtMetRS/BL21;2:Lysis solution of pET32a-MtMetRS/BL21 induced by IPTG;3:Recombinant MtMetRS expressed by pQE60-MtMetRS/M15;4:Lysis solution of pQE60-MtMetRS/M15 induced by IPTG;5: Recombinant MtMetRS expressed by pTrcHisB-MtMetRS;6: Lysis solution of pQE60-MtMetRS/M15 induced by IPTG;7: Recombinant MtMetRS expressed by pGEX-6P-1-MtMetRS/BL21;8: Lysis solution of pGEX-6P-1-MtMetRS/BL21 induced by IPTG圖3 重組蛋白的表達篩選Fig.3 Expression screening of recombinant protein
2.4MtMetRS重組蛋白的制備 pQE60-MtMetRS/M15接種到2 L LB培養中,18 ℃ 0.5 mmol/L IPTG過夜誘導表達。重組MtMetRS的分子排阻層析結果如圖4所示。MtMetRS的洗脫峰成對稱圖形,提示重組蛋白的結構均一。根據標準品的分子量及洗脫峰值計算獲得的方程式為Y=7.900 35-0.186 09X。重組MtMetRS的洗脫峰體積為16.86 mL,依據上述方程計算出分子排阻層析測量的重組蛋白分子量為57.93 kDa,與MtMetRS的理論分子量59.12 kDa近似。使用Nanodrop的測定值除以重組蛋白的消光系數1.371,調整蛋白質的濃度為10 mg/mL,50 μL液氮速凍后-80 ℃保存。
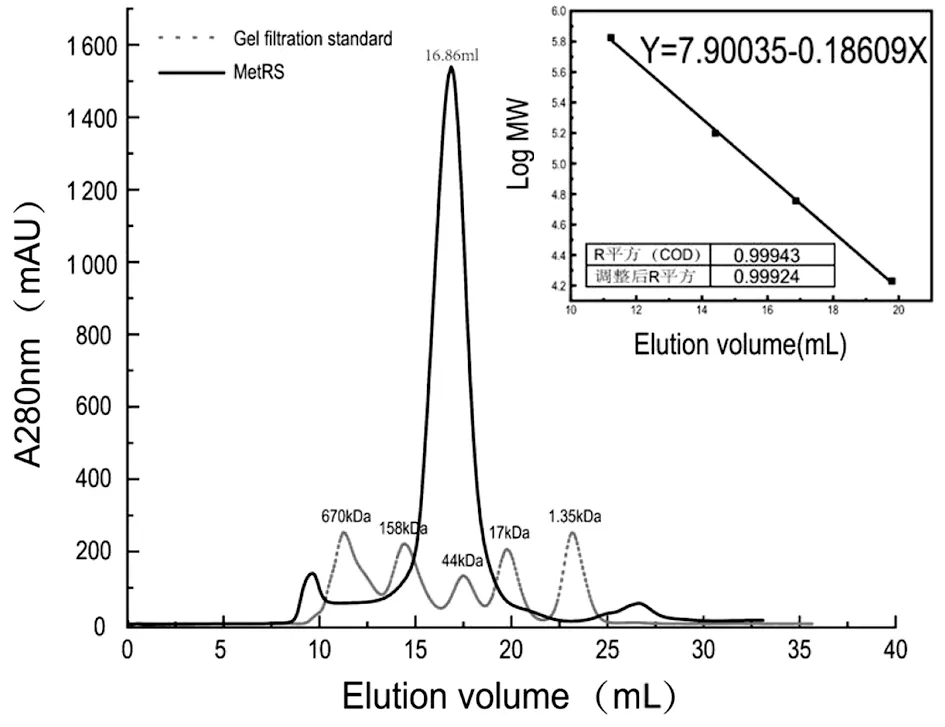
圖4 重組MtMetRS及標準品的gel filtration洗脫峰Fig.4 Gel filtration elution peaks of recombinant MtMetRS and standard

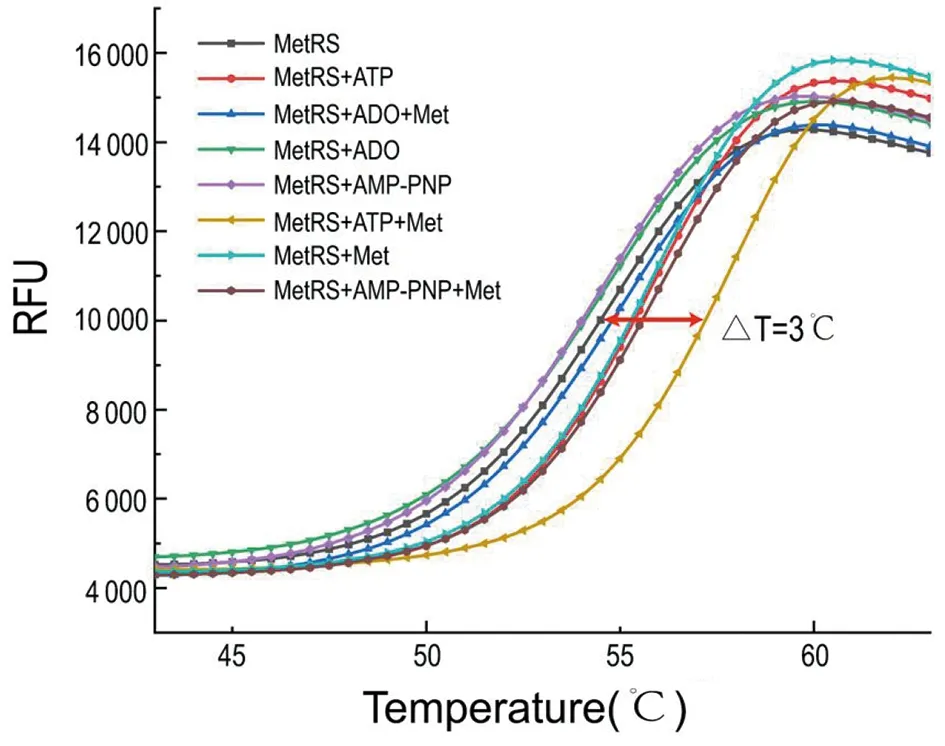
圖5 重組MtMetRS的TSA分析Fig.5 TSA to assess ligand:MtMetRS binding
為了進一步分析TSA的結果,我們在圖6中展示了利什曼原蟲及恥垢分枝桿菌MetRS與底物結合的晶體結構。從圖6A中可以看到Met結合區是一個疏水口袋,其通過疏水區域的大小和疏水特性識別和結合Met[3]。Met的這種結合方式對提高MetRS的熱穩定性貢獻有限。而MetRS與ADO和Met結合時,不能引起催化口袋的封閉,催化口袋處于開放狀態,如圖6A所示。只有MetRS與Met和ATP結合后,催化形成的MetAde和PPi可以引起催化loop的構象變化,封閉催化口袋,如圖6B所示。形成封閉的催化口袋可能是MtMetRS熱穩定性提高的基礎。
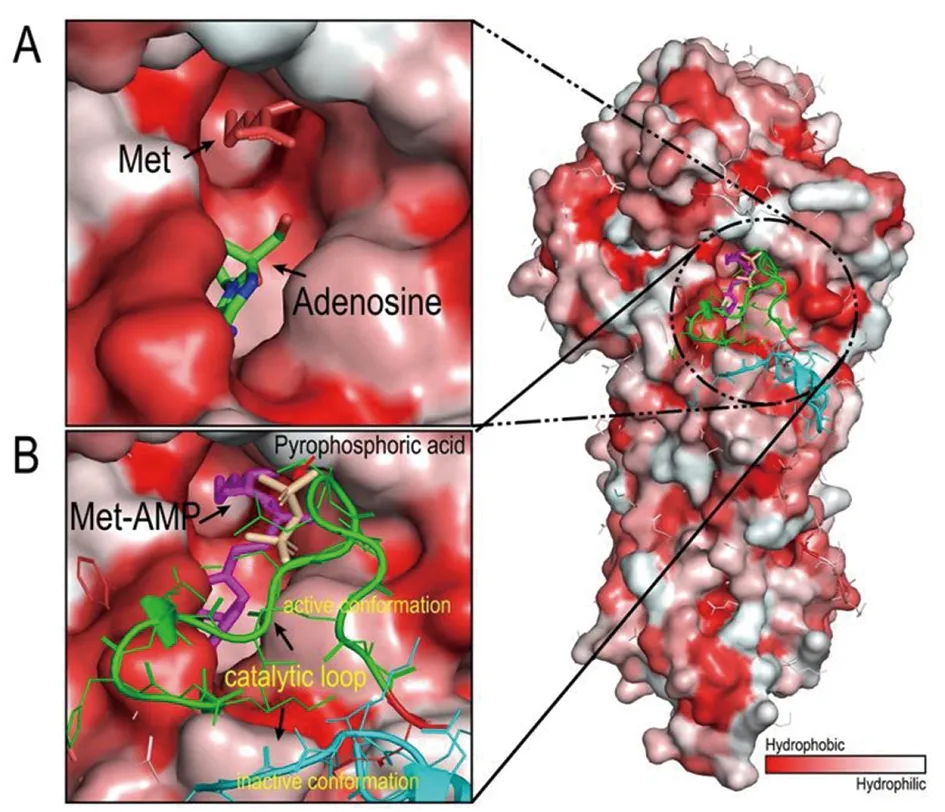
A: MetRS combined with Met and ADO(PDB:2X1L);B: MetRS combined with MetAde and PPi(PDB:3KFL)圖6 MetRS與配體結合的分子結構Fig.6 Molecular structure of MetRS and ligand binding
3 討 論
人們已經從天然來源或通過篩選合成文庫方法鑒定到了許多aaRS抑制劑[5-7]。甲硫氨酰tRNA合成酶與其他aaRS最大的區別是不同物種來源的MetRS顯示了結構的多樣性[8]。人們早就認識到MetRS作為藥物靶標的重要性,并不斷在MetRS抑制劑的研究中取得進展[9-16]。上個世紀90年代人們已經意識到MtMetRS在抗結核分支桿菌抑制劑研究中的重要性,初步報道了MtMetRS的部分生化參數[17]。但由于MtMetRS重組制備方法沒有突破,此后再沒有MtMetRS的研究報道。Ingvarsson等對MtMetRS晶體學的研究也是由于難以大量獲得重組MtMetRS而改為使用恥垢分枝桿菌MetRS為研究對像。
影響重組基因在原核細胞中表達的因素有密碼子頻率、蛋白質表達速度、目的蛋白的溶解性、mRNA結構等。在之前的MetRS研究報道中均使用原核表達系統獲得重組蛋白,因此我們認為密碼子頻率和蛋白的溶解性不是阻礙獲得重組MtMetRS的主要原因。pTrcHisB-MtMetRS質粒在插入基因的5′端存在一個小的順反子結構,可以改善質粒轉錄的mRNA 5′端結構,有利于mRNA與核糖體的結合,但是在表達篩選結果中沒有看出其明顯增加重組蛋白的表達。使用弱啟動子降低蛋白質表達速度可能是獲得重組MtMetRS的主要原因。
MetRS分子具有底物誘導的構象變化。在未結合底物時,催化口袋呈現開放狀態,而結合底物后催化口袋封閉。以往針對MetRS抑制劑的設計和篩選以開放狀態為對象,選擇化合物配體去結合催化口袋。我們的TSA分析結果和利什曼原蟲與MetAde結合模型顯示其催化口袋的封閉狀態對配體結合最強。這提示結合MetRS并促使其催化口袋封閉的化合物是很好的抑制劑。以往以MetRS催化口袋的開放狀態為目標設計、篩選抑制劑可能不是最優的方法。
總而言之,我們建立了重組MtMetRS的制備方法,為研究和篩選結核分枝桿菌抑制劑提供了基礎。同時,TSA結果更進一步為抗結核分枝桿菌抗生素的研究指明方向。
利益沖突:無
