天然礦物共混活性焦聯合低溫脫硫脫硝
2021-05-15 04:04:48楊林孟小謎姚露賴雨果蔣文舉
化工學報 2021年4期
楊林,孟小謎,姚露,賴雨果,蔣文舉
(1 四川大學建筑與環境學院,四川成都610065; 2 四川大學國家煙氣脫硫工程技術研究中心,四川成都610065)
引 言
在鋼鐵、有色冶金、建材等中低溫煙氣治理中,活性焦(activated coke,AC)脫硫脫硝工藝有著廣闊的應用前景[1-3]。活性焦具有能夠同時脫除煙氣中多種污染物的特點。然而,由于中低溫條件下,煙氣中的SO2、重金屬等物質容易導致活性焦快速中毒而失去脫硝活性,當前基于活性焦技術的大氣污染控制技術以及有關活性焦脫硫、脫硝研究主要是基于單一污染物的脫除[4-7]。
為解決上述問題,研究者提出采用先脫硫再脫硝的分段連續過程實現活性焦的聯合脫硫脫硝[8-9]。以常用的移動床反應器為例,具體設計方式如下:在反應器設計時將反應器劃分為兩個連續反應單元,待處理煙氣于第一反應單元內首先完成脫硫反應,然后經加熱后進入脫硝反應單元去除煙氣中的NOx。脫硝反應還原劑的添加設計在兩個反應單元之間。整個反應過程中,活性焦由反應器頂部進入,在自身重力的作用下與煙氣逆流接觸完成脫硝和脫硫反應,并在脫硫反應后從反應器底部排出進行再生處理。然而,常規活性焦普遍具有脫硝活性差和脫硫容量低的特點,導致該技術的投資和運行成本較高。
針對常規活性焦脫硫脫硝效率低的問題,近年來采用過渡金屬表面改性新型活性焦的研究得到極大關注[10-12]。常用的活性焦改性方法主要有浸漬法和共混法[1,13],共混法改性是在活性焦成型制備過程中將改性添0 加劑固體粉末直接與原料碳粉混合,改性劑通過參與活性焦的整個炭活化反應過程完成表面改性[13-15]。相比浸漬法,共混法改性將活性焦的制備與表面改性過程合二為一,操作簡單,改性制備成本較低。共混法改性也極大地拓展了改性劑的適用范圍,如過渡金屬氧化物、天然礦物精礦粉等均可用于活性焦的改性制備[10,16-17]。因此,以天然精礦粉或金屬氧化物為改性劑共混制備改性活性焦,將使活性焦制備成本顯著降低,進而推動活性焦脫硫脫硝工藝的工業應用進程。作者所在課題組在活性焦的共混改性及其脫硫和脫硝應用方面做了大量研究工作,特別是天然礦物共混改性在煙氣脫硫和脫硝中的應用[4,13,17-19]。
2008 年,Ma 等[20]以V2O5/AC 為催化劑研究了同時脫硫脫硝反應過程中V2O5對SO2脫除的作用。此后,Li 等[21]研究了多級流化床同時脫硫脫硝,發現4級流化床可以同時實現100%SO2和73.2%和NO 的去除。然而,上述研究或使用常規活性焦,或主要使用浸漬改性活性炭為研究對象,對于共混改性活性焦的聯合脫硫脫硝反應研究仍舊匱乏。本文在多種天然礦物共混改性活性焦脫硝研究的基礎上,以具有最佳脫硝活性的軟錳礦和鈦精礦復合共混改性活性焦為樣品,模擬研究基于移動床反應器的活性焦聯合脫硫脫硝反應,通過循環使用過程活性焦脫硫和脫硝性能測試,比表面積、表面金屬組分以及化學官能團等理化特性表征研究共混改性活性焦多過程連續反應的相互影響及作用,為推進活性焦一體化脫硫脫硝技術提供理論指導和技術支撐。
1 實驗材料和方法
1.1 材料
本研究活性焦的碳源為產自中國山西的煙煤和焦煤,黏結劑為四川省煤焦化集團有限公司提供的高溫煤焦油。改性劑為軟錳礦和鈦精礦,分別產自南非和中國攀枝花,基于XRF 分析得到的軟錳礦和鈦精礦組成如表1所示。
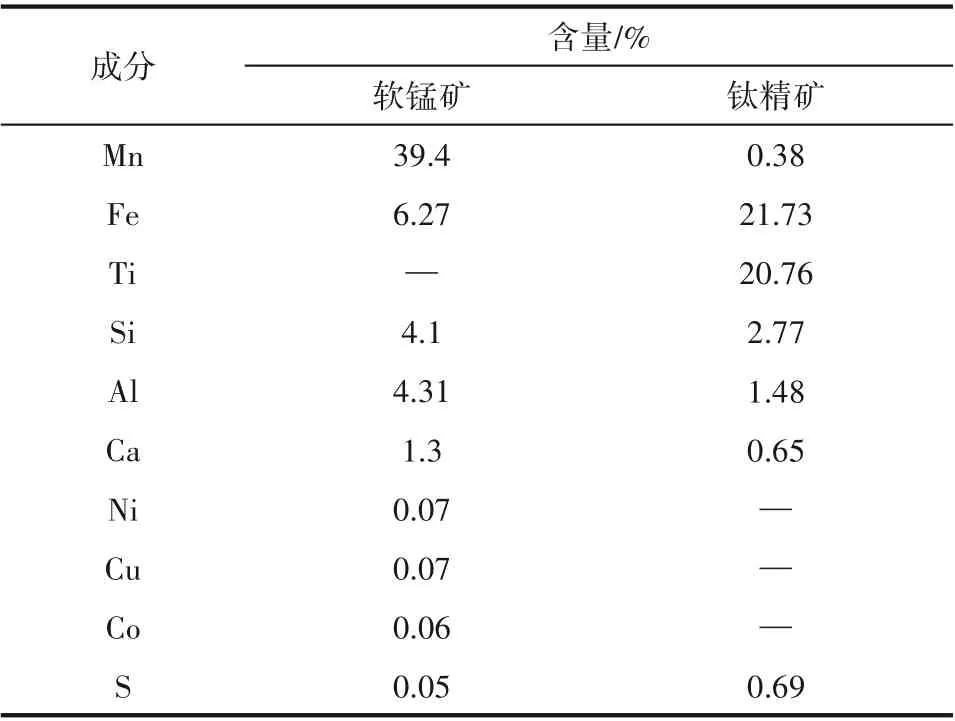
表1 軟錳礦和鈦精礦主要成分Table 1 The components of pyrolusite and titanium ore based on the XRF
1.2 活性焦的制備
本研究所用的活性焦為軟錳礦(pyrolusite,P)和鈦精礦(titanium ore, Ti)雙礦物共混改性活性焦。軟錳礦和鈦精礦按質量比為1/2、總添加量為15%(質量)共混制備活性焦,活性焦制備參考文獻[10],具體制備工藝如下:(1)將焦煤、煙煤及兩種天然礦物分別進行磨粉并過74 μm篩備用;(2)將煙煤和焦煤按照配比為7/3 混合,然后將總量為15%(質量)、質量比為1/2 的軟錳礦和鈦精礦分別依次加入煤粉并攪拌不低于30 min;(3)向攪拌均勻的粉料中按質量比加入10%的蒸餾水進行濕潤,然后再加入質量分數為40%的煤焦油,然后攪拌均勻至物料具有金屬光澤;(4)將混合物料轉移至模具中,采用陶瓷擠出機擠壓成型得到3 mm的柱狀半焦,得到的半焦經烘干后待用。半焦以水蒸氣為活化劑,采用一步法進行活化。碳活化處理時,首先升溫至600℃保持1 h進行碳化處理,然后進一步升溫至900℃恒溫停留1 h,期間按碳/水=2/1 均勻通入相應質量水蒸氣進行活化。活化結束后自然降溫至室溫。整個升溫過程采用5℃/min 升溫速率。所得活性焦命名為P1/Ti2-15@AC。其中,AC 表示活性焦,P1/Ti2 表示軟錳礦和鈦精礦的負載量之比為1/2,15 表示兩種天然礦物的總負載量。
1.3 活性焦聯合脫硫脫硝及再生
本研究采用脫硝-脫硫-再生連續循環過程模擬移動床反應器中活性焦聯合脫硫脫硝反應過程,該過程中,活性焦首先進行脫硝反應,脫硝反應完成后轉至脫硫活性測試,最后完成脫硫反應的活性焦進行再生處理,依次循環5次。脫硝反應條件為:NO = 670 mg/m3,O2=5%(體積),N2載氣,反應溫度150℃,反應空速為1000 h-1。脫硫反應條件為:SO2= 8570 mg/m3,O2= 8%~10%(體積),N2為載氣,反應溫度80℃,反應空速為600 h-1。活性評價反應器內徑為21 mm,催化劑的裝填高度為10 cm。反應過程中,煙氣進出口SO2和NO 濃度采用Gasboard-3000 在線紅外煙氣分析儀連續監測并采樣。脫硝反應NO轉化率的計算如式(1)所示:

式中,η 為NO 轉化率,%;Cin為進口NO 濃度,mg/m3;Cout為出口NO濃度,mg/m3。
活性焦的硫容計算如式(2)所示:
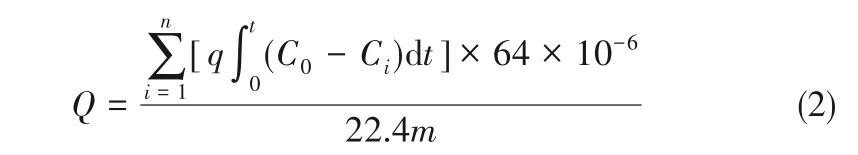
式中,Q 為活性焦硫容,mg/g;q為入口氣體總流量,ml/min;C0為入口SO2濃度,mg/m3;Ci為第i 次采樣出口SO2濃度,mg/m3;t 為第i 次采樣間隔時間,min;n為采樣次數;m為活性炭裝填量,g。
活性焦依次經過脫硝和脫硫反應后,轉移至再生裝置進行再生。熱再生條件為400℃,N2氛圍保護,恒溫保持60 min。熱再生過程產生的高濃度SO2氣體采用稀雙氧水吸收。為保證循環反應過程中樣品的均勻性,每個循環的脫硫反應結束時煙氣出口SO2濃度不低于進口濃度的90%。脫硫容量以10%穿透計算。再生后活性焦按照再生次數記為P1/Ti2-15@AC-Rn,其中Rn表示第n次再生。
1.4 材料表征
活性焦比表面積和孔結構特征分析基于恒溫氮氣吸脫附測試,所使用的儀器為美國麥克ASAP 2460 氣體吸附儀。活性焦的比表面積、微孔體積和中孔體積分別采用BET、t-plot和BJH 模型進行計算分析。XRD 分析采用X-Pert PRO MPD 型衍射儀,以Cu Kα為放射源,于30 V和20 A條件下進行掃描。測試掃描速率為0.02(°)/s,掃描范圍為10°~80°。FTIR 測試采用Nicolet 6700 型紅外光譜儀,用于獲得活性焦的表面碳氧官能團分布。FTIR 的掃描范圍為4000~400 cm-1,波數精度0.01 cm-1,分辨率0.09 cm-1。XPS 分析采用美國PHI 5000C ESCA system 能譜儀,以Al Kα(1486.60 eV)為射線源,工作電壓為12 kV,電流為15 mA。
2 結果與討論
2.1 連續脫硫脫硝性能
圖1(a)所示為P1/Ti2-15@AC 不同脫硝-脫硫-再生循環后的脫硫穿透曲線。如圖所示,再生后P1/Ti2-15@AC 的脫硫性能逐漸下降,100%全脫硫時間縮短。表2所示為不同再生次數后活性焦的硫容和10%穿透時間。P1/Ti2-15@AC 的穿透硫容和穿透時間分別為90.9 mg/g 和17.3 h。首次再生后,P1/Ti2-15@AC-R1 的硫容和穿透時間分別為75.9 mg/g 和14.0 h,相比新樣品分別減少了16.5%和19.2%。隨著再生次數的增加,硫容和脫硫時間持續縮減,至第五次再生后(P1/Ti2-15@AC-R5)活性焦的穿透硫容和穿透時間減少至僅為49.9 mg/g 和8.2 h。
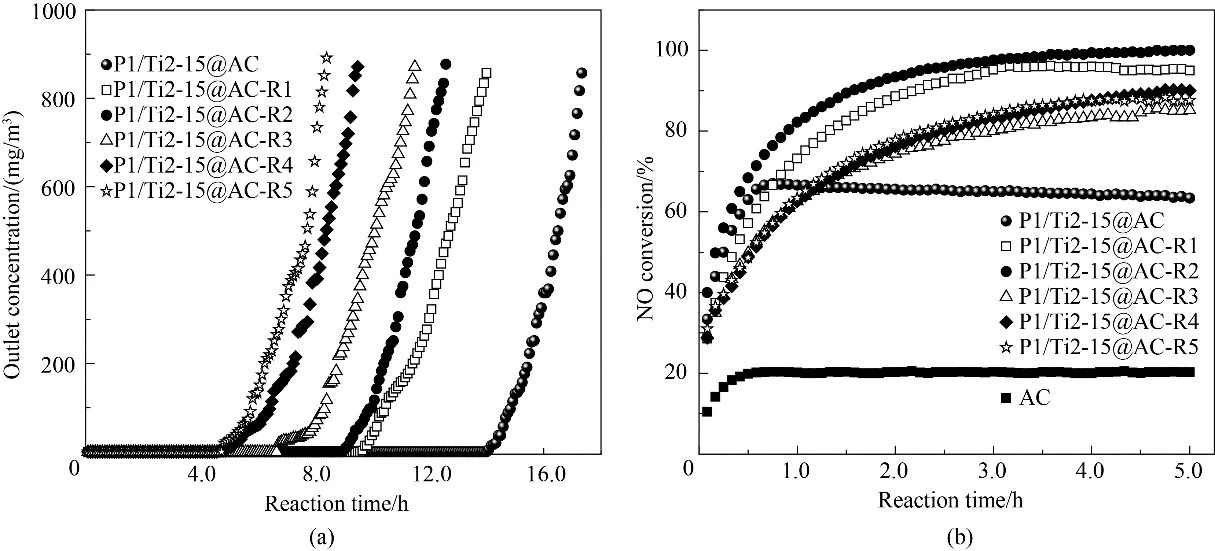
圖1 P1/Ti2-15@AC循環脫硝-脫硫-再生的脫硫穿透曲線(a)及脫硝反應曲線(b)Fig.1 The flue gas desulfurization(a)and denitrification(b)curves of the cyclic used P1/Ti2-15@AC
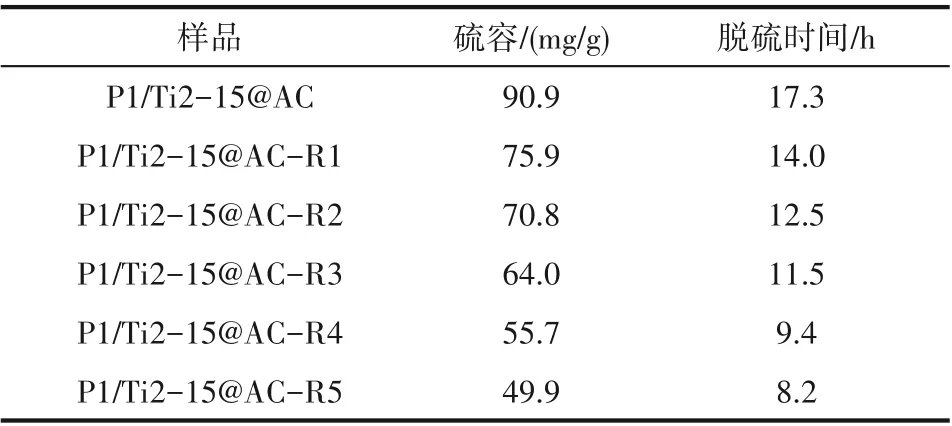
表2 P1/Ti2-15@AC循環脫硝-脫硫-再生的脫硫硫容及穿透時間Table 2 Sulfur capacities and breakthrough time of the cyclic used P1/Ti2-15@AC
圖1(b)所示為P1/Ti2-15@AC 不同循環次數后的脫硝NO 轉化率。如圖所示,新的P1/Ti2-15@AC在150℃時的NO 穩定轉化率為67.0%,該值明顯優于已報道過渡金屬改性活性炭的脫硝NO 轉化率[5]。經過首次脫硫反應和再生過程后,P1/Ti2-15@ACR1 的脫硝活性得到顯著提升,脫硝NO 轉化率升高至95.0%。二次再生后,P1/Ti2-15@AC-R2 的脫硝NO 轉化率達到最高的100.0%。隨著循環次數的繼續增加,P1/Ti2-15@AC-Rn 的脫硝活性在表現出一定程度的降低后保持基本穩定,第三至第五次循環再生后活性焦的脫硝NO 轉化率分別為85.2%、90.0%和87.6%。P1/Ti2-15@AC(-Rn)在反應初期均具有一個活性逐步升高的過程,這可能是由于活性焦載體具有較大的NH3吸附容量所導致的。上述結果表明,循環脫硝-脫硫-再生過程中,P1/Ti2-15@AC 的脫硫再生過程具有強化其脫硝反應活性的作用。以下將基于循環使用過程中活性焦表面理化特性表征進行具體分析。
2.2 連續脫硫脫硝過程中表面特性的變化
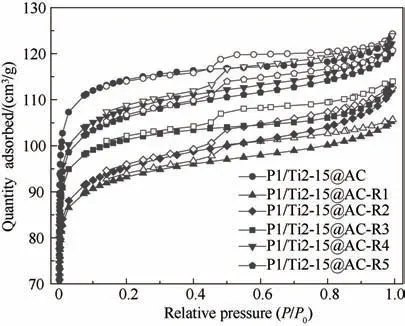
圖2 再生前后P1/Ti2-15@AC的N2吸-脫附曲線Fig.2 N2 adsorption-desorption curves of cyclic used P1/Ti2-15@AC
2.2.1 孔隙結構與比表面積 圖2 所示為不同脫硝-脫硫-再生循環次數后P1/Ti2-15@AC 的N2等溫吸脫附曲線。P1/Ti2-15@AC 和P1/Ti2-15@AC-Rn在相對壓力(P/P0)小于0.1 的區間內均表現出較高的N2吸附容量,表明再生前后活性焦均具有發達的微孔結構[22]。此外,所有分析樣品在中高壓段都有明顯的H4 型回滯環,表明其同時具有一定量的中孔 結 構[23]。表3 所 示 為P1/Ti2-15@AC 和P1/Ti2-15@AC-Rn 的比表面積及孔結構特征參數。P1/Ti2-15@AC 的比表面積和總孔體積分別為364 m2/g和0.192 cm3/g。首次脫硫再生后,P1/Ti2-15@ACR1的比表面積和孔體積相比P1/Ti2-15@AC 均出現了不同程度的降低。但是,P1/Ti2-15@AC-R1 中孔體積得到顯著增大,由P1/Ti2-15@AC 的0.024 cm3/g增大至0.031 cm3/g。微孔體積的減小主要有兩個原因,一是脫硫產生的硫酸鹽會堵塞一部分微孔,二是再生過程中硫酸與碳的反應導致部分微孔結構坍塌,這也是P1/Ti2-15@AC-R1 中孔體積增大的主要原因。從第二次再生開始,P1/Ti2-15@AC-Rn 的比表面積和孔體積隨著再生次數的增加表現出一定的增大趨勢,表明此后的再生過程中碳與硫酸的表面反應以產生新的多孔結構為主,這與已有關于多孔碳材料的再生研究結果是一致的[18,24]。
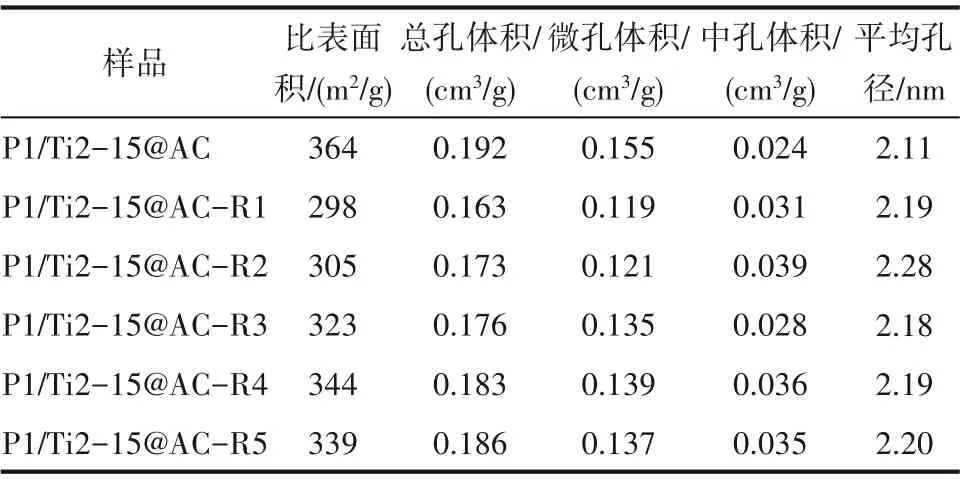
表3 再生前后P1/Ti2-15@AC的比表面積和孔隙結構參數Table 3 Textual properties of cyclic used P1/Ti2-15@AC
2.2.2 表面金屬形態分析 圖3所示為不同循環使用次數后活性焦的XRD 譜圖。如圖所示,P1/Ti2-15@AC 表 面 錳、鐵 和 鈦 分 別 主 要 以MnO(2θ=35.10°)/Mn3O4(2θ =41.58°)、Fe2O3(2θ =31.48° ,61.41°)和TiO2(2θ=31.48°,45.12°)的形式存在,同時檢測到MnFe2O4(2θ=56.16°,61.65°)和FeTiO3(2θ=48.87°)等雙金屬氧化物。相比于P1/Ti2 混合物,單一的過渡金屬氧化物在活化的高溫條件下實現了向多金屬復合氧化物的轉變,CaCO3發生分解,MnO2轉化為MnO。研究表明,Mn 在催化劑中以多種價態存在時可提高催化劑表面的氧化還原能力,有助于脫硝反應的進行[25]。鐵與錳形成雙金屬橋鍵結構,加速催化劑表面晶格氧的轉移和恢復,同時強化脫硝反應過程中的電子轉移[26-27]。TiO2主要是通過增加表面酸性位,加快脫硝反應過程中NH3的表面吸附速率。
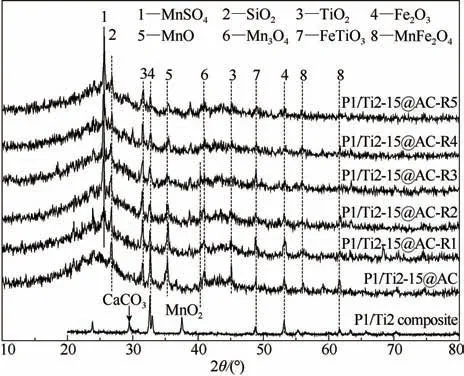
圖3 再生前后P1/Ti2-15@AC的XRD分析譜圖Fig.3 XRD patterns of the cyclic used P1/Ti2-15@AC
經過脫硫和再生反應后,P1/Ti2-15@AC-Rn 表面歸屬于MnO 和Fe2O3的特征峰逐漸減弱,而歸屬于TiO2的特征峰保持不變。再生后,P1/Ti2-15@AC-Rn表面出現了歸屬于MnSO4(2θ=25.67°)的新的特征峰,表明MnO 在脫硫過程中被逐漸轉化。Fe2O3的酸穩定性也較差,P1/Ti2-15@AC-Rn表面未檢測到硫酸鐵鹽可能是因為其較高的分散性和較差的結晶程度[13]。脫硫再生過程同時也破壞了活性焦表面雙金屬橋鍵結構,歸屬于FeTiO3和MnFe2O4的特征峰在脫硫再生后呈現不同程度的減弱,且隨循環次數的增多部分特征峰逐漸消失。由于金屬硫酸鹽的分解溫度普遍高于400℃,再生過程中活性焦表面的MnSO4等并不會發生分解,歸屬于硫酸鹽的特征峰強度也隨著循環使用次數的增加不斷增強。
2.2.3 表面官能團 圖4所示為不同循環使用次數后活性焦的紅外光譜圖。活性焦位于3440 cm-1的吸收峰歸屬于O—H 的伸縮振動特征峰,表明活性焦表面含有醇、羧酸或酚類官能團[28]。位于1460、1080 和875 cm-1的吸收峰分別對應的是CH2/CH3、COOH 和C—H[29]。隨著循環使用次數的增加,P1/Ti2-15@AC-Rn 位于1080 cm-1處的吸收峰相比P1/Ti2-15@AC 均表現出一定程度的增強,而位于1460和875 cm-1處的CH2/CH3和C—H的峰迅速減弱至消失。相比P1/Ti2-15@AC,再生后樣品在1580 cm-1和675 cm-1附近均出現了新的特征峰,它們分別是歸屬于醌基、羰基和芳香環中的C O/C C 振動疊加[30]和表面C—S磺酸官能團[31]。這些都是典型的酸性官能團,它們的增多可以有效改善脫硝反應過程中表面NH3的吸附過程,從而提高活性焦脫硝反應的NO 轉化率[32]。這些變化與增強的脫硝反應活性是一致的。
表4 所示為基于活性焦X 射線能譜分析中高分辨率C1s 譜圖,使用XPSpeak 軟件擬合解析得到的表面碳氧官能團分布統計數據[33]。相較于P1/Ti2-15@AC,P1/Ti2-15@AC-Rn 表面的含氧官能團均表現出不同程度的增加。P1/Ti2-15@AC-R1 具有最多的表面含氧官能團,然后隨著循環再生次數的增加不斷減少。對于炭基材料,C O和離域π電子主要顯堿性[34],C—O 官能團主要顯酸性[35]。代表活性焦表面酸性的C—O 含量的增加,表明再生樣品較未再生時有更好的脫硝性能。離域π電子碳含量的下降,表明再生樣品表面堿性基團的減少,從而導致脫硫性能出現下降。
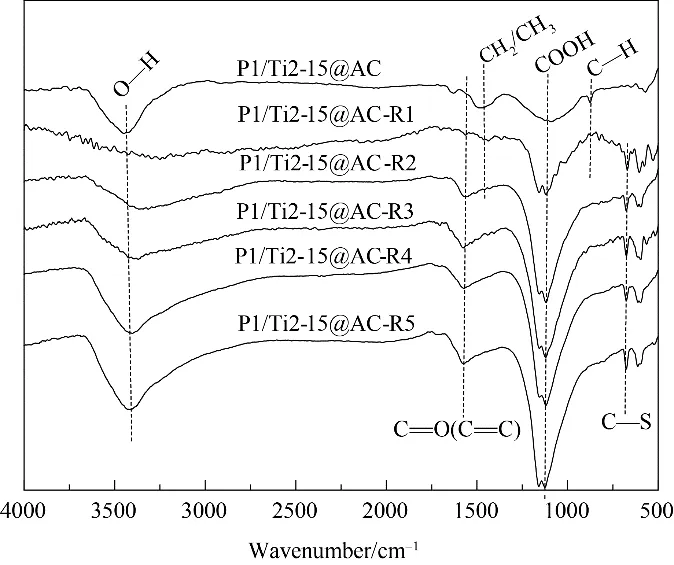
圖4 再生前后P1/Ti2-15@AC的傅里葉紅外譜圖Fig.4 FTIR spectra of the cyclic used P1/Ti2-15@AC
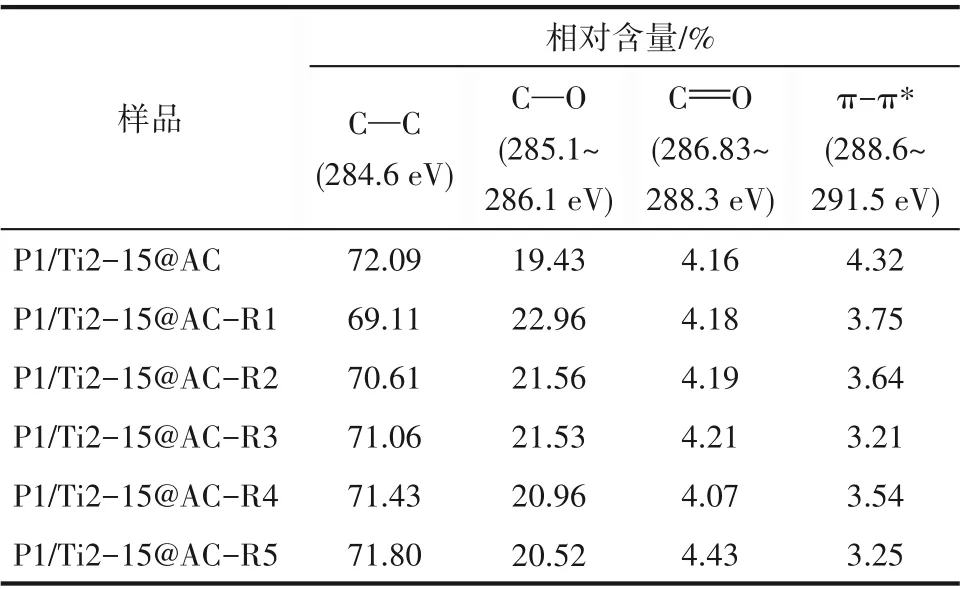
表4 基于C 1s譜圖所得P1/Ti2-15@AC不同循環次數后表面碳氧官能團的分布Table 4 Binding energy and relative content of cyclic used P1/Ti2-15@AC(based on the C1s spectrum)
2.2.4 表面硫元素分析 圖5 所示為循環使用后P1/Ti2-15@AC-Rn 表面S 2p 窄譜掃描圖。如圖所示,再生后P1/Ti2-15@AC-Rn(n=1, 2, 3, 4, 5)的S 2p 譜圖分別在169.4、169.4、169.4、169.2、169.1 eV處出現一個獨立的特征峰。研究表明,S 2p 譜圖中位于169.0~167.0 eV 之間的特征峰是歸屬于硫酸根結構中的硫[36-37],表明再生后活性焦表面的硫主要以硫酸根形式存在,這與XRD 得到表面大量硫酸鹽存在的結果一致。而在紅外光譜分析中P1/Ti2-15@AC-Rn 表面檢測到了C—S磺酸官能團,這可能是由于活性焦表面C—S 磺酸官能團分布不均所導致的。
2.3 連續脫硫脫硝綜合分析
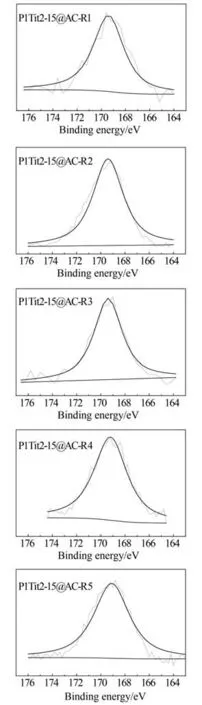
圖5 再生后P1/Ti2-15@AC表面S 2p圖譜Fig.5 S 2p XPS peak of the cyclic used P1/Ti2-15@AC
在P1/Ti2-15@AC 聯合脫硫脫硝循環使用過程中,脫硫和再生過程通過顯著改變活性焦的表面官能團進而影響其脫硫脫硝性能。隨著再生次數的增加改性活性焦的脫硫性能降低,這是因為活性焦微孔結構和表面堿性官能團對其脫硫活性至關重要。首先,再生后改性活性焦的比表面積較新樣品均有不同程度的降低,且再生后活性焦的微孔體積明顯減少;其次,再生后活性焦表面離域π電子等顯堿性官能團含量減少;再次,隨著脫硫反應的持續,活性焦表面具有脫硫催化活性的金屬氧化物的不斷消耗。上述多種變化共同作用導致活性焦脫硫性能逐漸減弱。
而相比于P1/Ti2-15@AC,P1/Ti2-15@AC-Rn脫硝性能得到顯著提升的主要原因是脫硫再生后活性焦表面酸性官能團增加,包括C—O、—COOH、硫酸鹽以及再生表面新生成的C—S 磺酸官能團[38-39]。酸性位的增加主要是通過改善脫硝過程中的NH3吸附過程,進而促進了SCR 反應的進行[40-42]。此外,也有研究表明硫酸錳和氧化錳共存可以增強活性焦的脫硝活性[43-44]。
3 結 論
在P1/Ti2-15@AC 聯合脫硫脫硝循環過程中,由于表面金屬氧化物和堿性官能團等活性位逐漸被消耗,P1/Ti2-15@AC-Rn 的脫硫性能逐漸降低。但是,活性焦經過脫硫再生反應后表面酸性官能團含量增加,脫硝反應過程中NH3的吸附得到改善,結合表面過渡金屬組分催化反應活性,使再生后活性焦脫硝活性都得到顯著提升。上述研究結果表明,采用新型天然礦物共混改性活性焦,結合加熱再生設計,可以很好地將煙氣脫硫和脫硝過程結合,從而實現高效率、低成本的聯合一體化脫硫脫硝。
猜你喜歡
化工管理(2022年13期)2022-12-02 09:21:52
山東冶金(2019年2期)2019-05-11 09:12:16
測控技術(2018年2期)2018-12-09 09:00:52
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年5期)2016-04-16 05:25:36
當代化工研究(2016年9期)2016-03-20 16:22:15
中國資源綜合利用(2016年2期)2016-01-22 07:27:41
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
