脯氨酰羥化酶抑制劑治療腎性貧血的研究進展
2021-05-21 13:05:30鄭守軍徐浩銅
攀枝花學院學報 2021年2期
關鍵詞:水平
鄭守軍,魏 巍,徐浩銅
(攀枝花學院 醫學院,四川 攀枝花 617000)
慢性腎臟病 (chronic kidney disease, CKD) 已對全球公共衛生構成巨大威脅,在我國大約有1.2億CKD患者,并且在成人人口中發病率高達10.8%[1-2]。貧血是CKD患者的一種常見并發癥,且隨腎功能下降發生率增高。據調研,我國CKD透析患者貧血患病率高達98.2%,非透析患者貧血患病率為52.1%[3-5]。目前,貧血癥的治療手段包括注射紅細胞生成刺激劑 (erythropoiesis stimulating agents, ESAs)、補鐵和輸血[6]。這些治療方式雖然能夠改善患者的貧血,但是有些不良反應限制了它們的臨床應用:ESAs能使體內半合成超生理劑量的促紅細胞生成素 (EPO),這讓卒中、死亡、充血性心力和心肌梗死衰竭一系列嚴重不良反應的發病風險增加,同時也讓超敏反應、癲癇和高血壓的發病風險上升[7];多數患者在服用鐵劑后會出現嚴重的胃腸道反應甚至過敏,而且鐵劑補充鐵的量通常會超過人體的負荷,使人體內的鐵過載和非轉鐵蛋白結合鐵增加,從而導致氧化損傷的擴大和增加感染風險[5];輸血會引發溶血反應、過敏、發燒、急性肺損傷甚至會造成疾病傳播和血液污染等[5]。因此,臨床上需要開發新的療法,以減少對CKD貧血的輸血依賴性,改善患者的生活質量,避免導致與EPO相關的不良反應。近幾年,國內外研究者開始探索新型的紅細胞生成刺激劑—低氧誘導因子脯氨酰羥化酶 (hypoxia-inducible factor-prolyl hydroxylase, HIF-PHD) 抑制劑。
1 低氧誘導因子脯氨酰羥化酶抑制劑簡介
1.1 低氧誘導因子 (HIF) 結構及生物學功能
HIF是體細胞在低氧濃度下啟動的一種轉錄因子,廣泛分布于血管內膜、心臟、腦、腎臟、肝臟等部分,調控EPO、血管內皮生長因子 (VEGF) 以及糖酵解酶等基因的轉錄[8-9]。HIF由HIF-α和HIF-β構成[10],這兩種亞基的氨基端都存在基本螺旋-環-螺旋(basic-helix-loop-helix) 和Per-ARNT-Sim (PAS)兩種結構域,這對形成異二聚體并與DNA結合是必不可少的[9]。作為活性亞基的HIF-α,基因定位于人的14號染色體q21~24區,含有826個氨基酸,感受缺氧信號的活性調控區域在其兩個末端,脯氨酸-絲氨酸-蘇氨酸 (Pro/Ser/Thr) 的氧依賴降解結構域 (oxygen-dependent degradationdomain, ODDD) 和轉錄激活結構域 (transactivation domain, TAD-C)廣泛存在于C末端,N末端具有TAD-N,這些結構域都是缺氧誘導蛋白轉錄激活、穩定和核定位的調控域,且TAD-C調整作用相對其他結構域更加精細,具有TAD-N是激活轉錄的必要條件。基因定位于人的1號染色體q21區的HIF-β又名芳香烴受體核轉運子 (aryl hydrocarbon reeptor nuclear translocator, ARNT),是組成性亞基,不受氧濃度影響,多數情況下其表達過量[11]。
HIF-α是氧高度敏感因子,有三種亞型:HIF-1α、HIF-2α、HIF-3α,HIF-β與以上三種亞型之一組合都能誘導靶基因的不同表達[12]。HIF-1α在絕大多數組織中均有表達,對造血干細胞細胞周期調節起關鍵作用,是治療缺血性疾病的潛在靶點,敲除HIF-1α基因的胚胎小鼠會因嚴重的心血管缺陷死亡。HIF-2α僅在特異細胞中表達,是低氧應答過程的主要介導者,參與上調EPO基因表達和低氧鐵轉運等。抑制HIF-1α,將導致HIF-2α量增加,從而轉錄功能增強,因此兩者在功能上存在差異但又互相補充。DNA結合域在HIF-3α中不存在,因此不會影響基因表達,但其可以通過HIF介導的基因表達,對HIF-1α和HIF-2α的產生抑制[12-14]。
1.2 脯氨酰羥化酶 (PHD) 的結構及生物學功能
PHD屬于2-酮戊二酸 (2-oxogluarate, 2-OG) (結構式見圖1) 和Fe2+依賴性雙加氧酶,在有氧情況下催化HIF-α特定脯氨酸殘基羥基化,包括PHD1、PHD2和PHD3和PHD4四種亞型:PHD1分布在細胞核中,PHD2分布在細胞質中,PHD3在細胞核和細胞質中都有分布,PHD4主要分布在內質網中[15]。
研究表明,PHD4只在HIF-1α過表達時發揮其調節作用[16];PHD1~3因組織分布不同 (表1),功能也各不相同:PHD1~3均能羥基化HIF-α的脯氨酸殘基,但其活性強度為:PHD2>>PHD3>PHD1。有研究顯示,抑制PHD2可促進HIF-2α聚集,從而內源性EPO水平提高,產生的EPO是腎臟中EPO的主要來源;而PHD1和PHD3在肝臟中也對氧依賴的EPO基因轉錄起到一定作用[17]。PHD1基因敲除小鼠看上去正常,但其從氧化代謝轉變為厭氧代謝:骨骼肌線粒體耗氧量減少,對缺血的耐受性增強,并且在運動測試中表現較差[18];PHD2基因敲除小鼠胚胎因心臟和胎盤異常導致死亡且顯示血管和紅細胞生成增加[19];PHD3基因敲除小鼠表現出明顯的交感神經功能變化[20]。
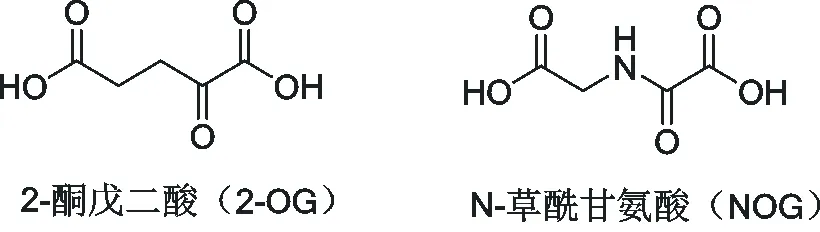
圖1 2-OG和NOG結構式

表1 PHD特性及常氧狀態下的組織分布

表1(續)
1.3 脯氨酰羥化酶 (PHD)/希佩爾-林道病腫瘤抑制蛋白 (pVHL) 信號通路對HIF-α的調節
在O2、Fe2+以及2-OG存在的條件下,PHD可實現多種底物 (如HIF-α、NF-κB、激活轉錄因子等) 的羥基化。正常氧濃度下,PHD可以識別HIF-α的脯氨酸殘基(HIF-1α: Pro402和Pro564, HIF-2α: Pro405和Pro531, HIF-3α: Pro409) 使其羥基化,經希佩爾-林道病腫瘤抑制蛋白 (pVHL) 介導,通過E3連接酶復合物泛素化途徑快速降解[14];在缺氧條件下,PHD活性下降,未被羥基化的HIF-α轉移到細胞核內,與HIF-β結合形成活性轉錄因子二聚體,再與轉錄共激活因子p300/Creb結合蛋白 (p300/CBP) 結合,使HIF信號通路激活,啟動下游EPO轉錄,增加血漿中EPO水平,刺激骨髓中紅細胞生成[21]。同時,低氧環境在激活HIF信號通路后,使得肝臟鐵調素合成受抑制,轉鐵蛋白增加,從而促進鐵的攝取和利用 (圖2)[22-23]。
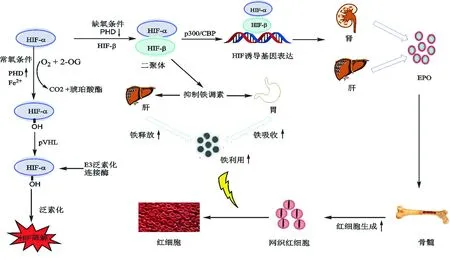
圖2 PHD/pVHL信號通路對HIF-α的調節示意圖
1.4 HIF-α其他調節通路
1.4.1 HIF-α上游調節通路
1.4.1.1 磷脂酰肌醇3-激酶 (PI3K) /蛋白激酶B (PKB/AKT) /哺乳動物雷帕霉素靶蛋白 (mTOR) 信號通路對HIF-α的調節
缺氧以及各種生長因子和細胞因子均可上調HIF-α蛋白表達,促進HIF-α靶基因表達,同時這些細胞因子可與相應的酪氨酸酶受體結合,激活PI3K、ATK以及mTOR信號轉導通路[24]。AKT磷酸化活化后能促進叉頭狀轉錄因子家族、正性調節核因子κB和CREBP以及促凋亡分子BAD (Bcl-2 /Bcl-xL associated death promoter) 磷酸化,增強抗凋亡基因轉錄,增強抗凋亡因子蛋白活性、降低凋亡因子表達[25],AKT的過度磷酸化能夠使細胞生長繁殖增快[26]。mTOR的C端與PI3K催化結構域同源, AKT活化后可促進mTOR的表達,高表達的mTOR可抑制AKT活性,mTOR對細胞增殖、凋亡以及成瘤具有重要調節作用,是磷酸化AKT下游重要效應分子[27]。mTOR通過5’端寡聚嘧啶核苷酸序列促使HIF-α mRNA翻譯速率增快,從而促進HIF-α表達[28]。
1.4.1.2 細胞外信號調節激酶 (ERK)/絲裂原活化蛋白激酶 (MAPK) 信號通路對HIF-α的調節
ERK/MAPK信號通路在生物調節過程中起著至關重要作用,例如細胞的增殖、分化、組織侵襲及轉移。MAPK路徑包含MAPK激酶激酶、MAPK激酶及MAPK三個主要激酶,ERK1,2為低進化的廣泛存在的絲氨酸蘇氨酸激酶,其在正常和病理狀態下調節細胞信號,ERK的過度表達在腫瘤的發生發展中其了至關重要的作用。Ras/Raf/MAPK(MEK)/ERK是MAPK相關通路中最為重要的信號聯級,Ras激活后通過該信號聯級激活下游蛋白,HIF-α是諸多ERK/MAPK信號通路調節靶點中的重要成員[29]。體外實驗證明HIF-α通過p42、p44、p38α和p38γ激酶使自身磷酸化來顯著增強活性,但HIF-α活性增強后并沒有明顯增加HIF-α蛋白的表達量[30]。ERK/MAPK信號能增強HIF-α蛋白活性,調節相應基因及蛋白的表達,并最終引起細胞增殖、凋亡、侵襲等相關生物學變化。
1.4.1.3 Bcl-2相關性抗凋亡基因3 (BAG3)/熱激酶蛋白70 (HSP70)蛋白酶體途徑對HIF-α的調節
BAG3隸屬于BAG家族,它與HSP70結合成蛋白酶體,作用是促進線粒體細胞凋亡。BAG3蛋白的表達大多發生在胞質,胞質中的HSP70蛋白上有BAG3蛋白結合位點,BAG3與胞質中的HSP70相對應的位點結合,進而促使HSP70與Bax (Bcl-2 associated X protein) 蛋白結合,最終形成復合體(BAG3-HSP70-Bax),這樣可使細胞中游離狀態的Bax蛋白含量降低;由于細胞線粒體附著有Bax蛋白,Bax蛋白含量降低可導致線粒體膜電位降低和線粒體通透性的增強從而加速線粒體裂解,最終導致細胞凋亡[31]。HSP70蛋白能通過20S和26S蛋白酶體促進HIF-α蛋白降解[32],而BAG3可與HSP70形成蛋白酶體復合物,這樣可減少HSP70蛋白量,從而抑制其對HIF-α的降解作用,以此促進HIF-α表達[33]。體外研究表明,非特異性蛋白酶體抑制劑MG132可使腫瘤細胞中HIF-α表達量升高[34],進一步證明了BAG3-HSP70蛋白酶體途徑抑制HSP70對HIF-α的降解作用。
1.4.2 HIF下游調節通路
調控靶基因的轉錄是HIF-α的下游調節的主要途徑,目前發現,血管內皮生長因子(vascular endothelial growth factor,VEGF)、血小板衍生生長因子(Platelet derived growth factor,PDGF)、葡萄糖轉運蛋白1(glucose transporter 1)、促紅細胞生成素(EPO)、內皮素1(Endothelin 1)、NO合酶2等均為HIF-α下游的靶基因,這些靶基因含有共同的啟動因子HRE,HIF-α蛋白能與靶基因HER序列特異性結合,對相應靶基因的轉錄起到促進作用[35]。研究證明,在缺氧條件下HIF-α靶基因表達量隨HIF-α表達增加而明顯增加[36]。
2 小分子口服HIF-PHD抑制劑研究進展
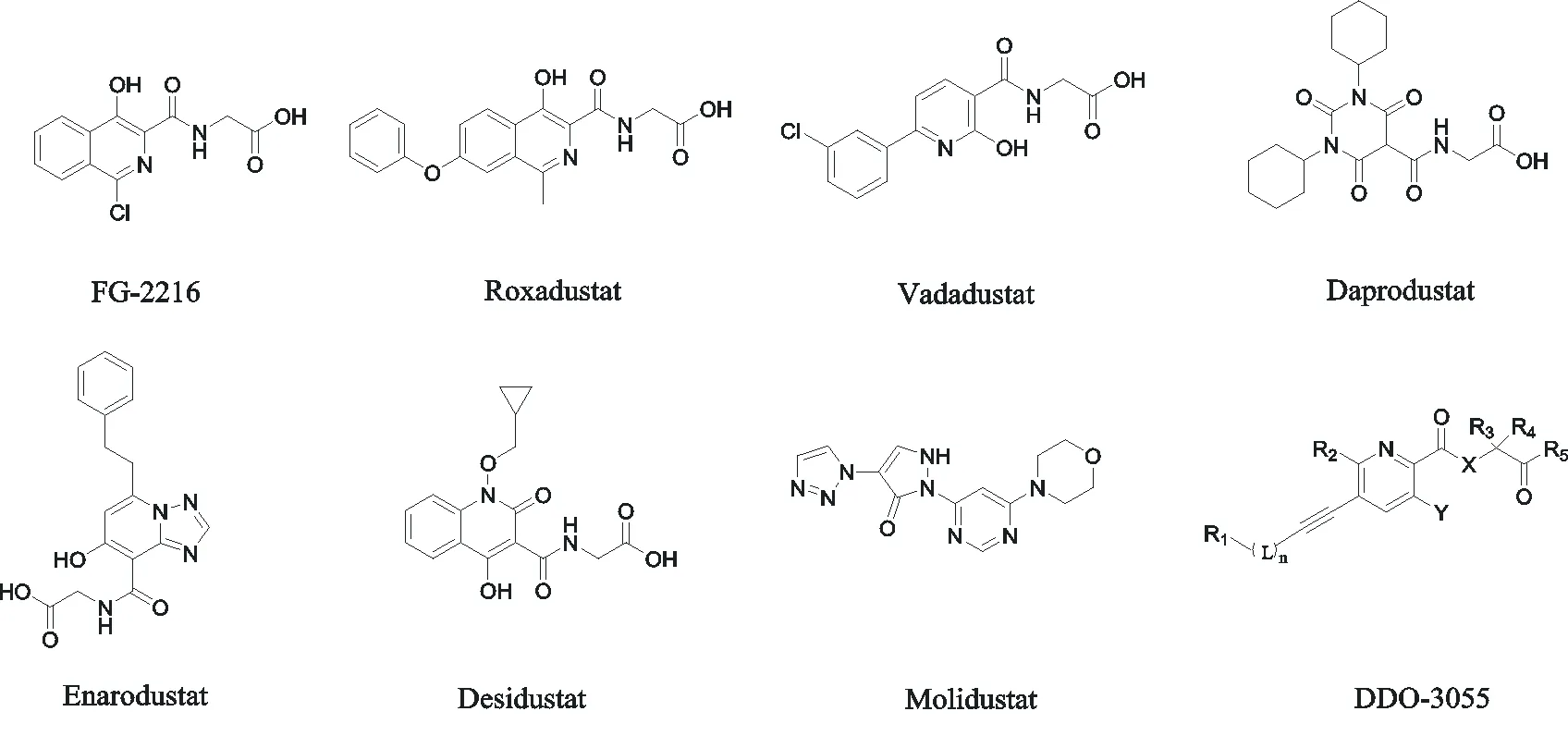
圖3 小分子HIF-PHD抑制劑
2.1 HIF-PHD抑制劑的結構特點
HIF-PHD屬于2-OG加氧酶,其作用是將氧分子中的氧原子直接引入其底物中[37]。N-草酰甘氨酸 (N-oxalylglycine, NOG) (見圖1) 與2-OG具有結構和極性相似,NOG類似物代替2-OG與Fe2+形成雙配位結合,由于其結合力相比2-OG更強,同時NOG酰胺結構降低了鄰位羧基碳原子的電子云密度降低,增加了氧親核進攻的難度,故使得PHD不能羥基化HIF,從而達到抑制HIF-PHD的效果[12]。目前國內外已上市或在研小分子HIF-PHD抑制劑結構式見圖3,由圖3可見FG-2216、羅沙司他、Daprodustat、Vadadustat、Enarodustat、Desidustat都保留了NOG側鏈。
2.2 小分子HIF-PHD抑制劑
HIF-PHD抑制劑通過抑制PHD來穩定HIF、刺激內源性EPO產生、上調轉鐵蛋白受體表達、增加鐵吸收、促進紅細胞成熟的小分子藥物[12,14],可以有效治療和預防HIF和EPO相關病癥,如貧血、局部缺血和缺氧的病癥。HIF-PHD抑制劑給CKD貧血治療帶來革命性的改變,目前僅羅沙司他已在中國上市,其余藥物都在臨床研究階段。下面將對目前已上市和在研的主要HIF-PHD抑制劑的研究進展和臨床療效進行簡要介紹 (表2)。
2.2.1 FG-2216
FG-2216是FibroGen公司開發的第一代HIF-PHD抑制劑,用于鐮狀細胞血癥、腎性貧血、化療誘導的貧血在內的各種貧血的潛在治療。1999年5月,FG-2216作為第一種口服HIF-PHD抑制劑進入臨床試驗階段[38]。在不給予ESAs治療的情況下,FG-2216能夠促進EPO基因表達上調,增加內源性EPO水平,其對PHD2的IC50為3.9 μmol/L,半衰期為15.7±1.1 h。但在Ⅱ期臨床研究中出現一例暴發性肝炎致死,隨即FDA全面暫停該藥物的臨床試驗,后期證明該死亡病例為非藥物引起,在2008年該藥物的臨床研究再次獲得FDA 批準,但目前并無進一步臨床研究報道。
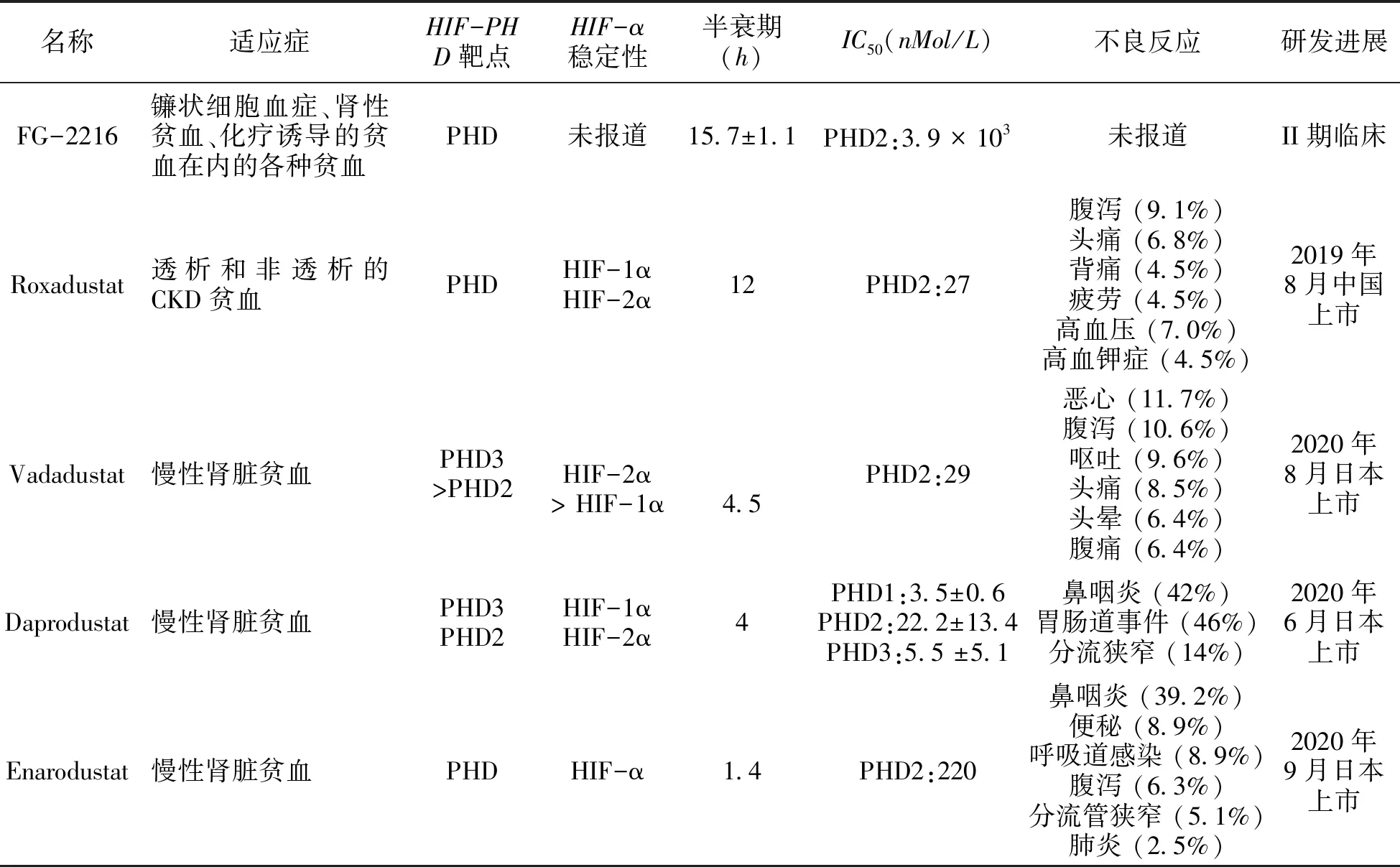
表2 小分子HIF-PHD抑制劑治療CKD貧血的研究進展
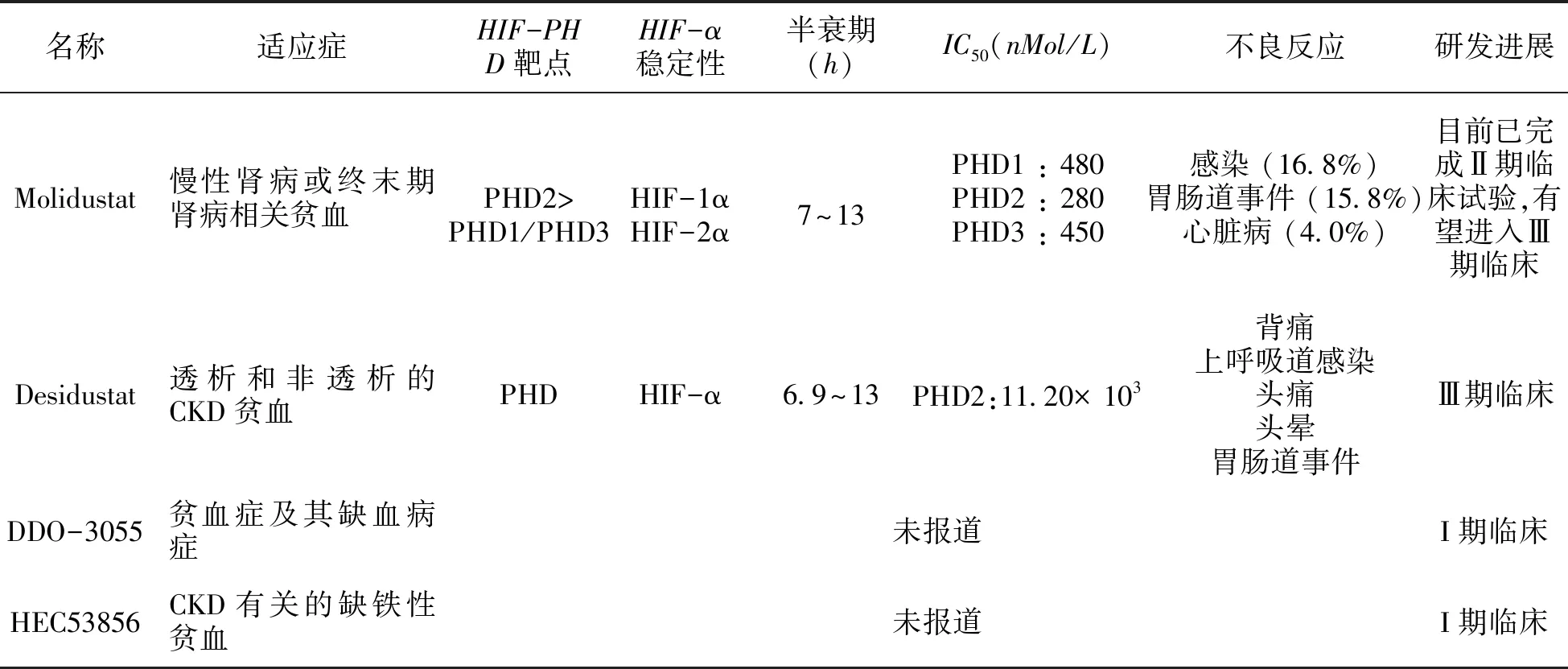
表2(續)
2.2.2 羅沙司他 (Roxadustat)
羅沙司他 (FG-4592) 是由FibroGen,Astellas Pharma和AstraZeneca合作開發的4-羥基異喹啉類化合物。2018年12月,NMPA以優先審評審批通過羅沙司他 (商品名:愛瑞卓) 用于治療正在接受透析的CKD貧血患者。2019年8月,羅沙司他非透析依賴性CKD貧血治療的新適應癥獲批。羅沙司他是中國率先獲批上市的first in class新藥,療效不劣于現有標準療法且具有口服的便捷性。
羅沙司他是第二代HIF-PHD抑制劑,與FG-2216相比,藥動學和藥效學有所改進。羅沙司他血漿半衰期約12 小時,臨床研究中的給藥方式是2-3次/周,每次口服1-2 mg/Kg[19],它能短暫、可逆的穩定HIF-α,使機體在不缺氧的前提下轉錄出相關基因,降低鐵調素水平,升高內源性EPO水平,上調轉鐵蛋白受體表達、增加鐵吸收、促進紅細胞成熟,從而發揮治療貧血的作用[39-40]。
中國開展的兩項III期臨床研究比較研究了羅沙司他對長期透析和未透析CKD貧血患者的療效和安全性,305例長期透析CKD貧血終末期患者中與對照組(阿法依泊汀)相比,羅沙司他對透析患者的臨床獲益和副作用沒有顯著差別[41];羅沙司他對154例未透析的CKD貧血患者有顯著的臨床療效,能有效糾正和維持CKD貧血患者Hb水平,且安全性和耐受性良好[42]。羅沙司他常見的不良反應為腹瀉、頭痛、背痛和疲勞;另外臨床試驗中有7%的出現了高血壓,雖然遠低于ESAs組[43]
2.2.3 Daprodustat
Daprodustat (GSK1278863) 是GSK公司研發的單環吡啶酮衍生物,已在日本和高加索地區完成臨床Ⅲ期研究。2020年6月,GSK在日本批準Daprodustat用于治療腎性貧血的新藥上市申請。
Daprodustat對PHD 3種亞型的半抑制濃度IC50分別為PHD1: 3.50.6 nM; PHD2: 22.213.4 nM; PHD3: 5.5 5.1nM,其主要通過CYP2C8在肝臟中代謝,半衰期為4小時。Ⅱ期臨床研究結果顯示,Daprodustat組內源性EPO造成的Hb水平升高顯著低于ESAs組,而且Daprodustat安全耐受性良好,血漿VEGF濃度沒有顯著升高;Daprodustat還可導致總膽固醇、低密度脂蛋白和高密度脂蛋白降低[7]。III期臨床研究納入271例依賴透析的患者進行了為期52周的研究,結果顯示口服Daprodustat在40到52周內的平均Hb水平上達到了阿法依泊汀靜脈注射液的非劣效性主要終點,在Daprodustat治療組中發生至少一種不良事件的患者百分比為93%,在對照組中為97%。治療組與對照組常見的不良事件是鼻咽炎 (42% vs 49%),胃腸道事件 (46% vs 46%) 和分流狹窄 (14% vs 15%)[44]。
2.2.4 Vadadustat
Vadadustat (AKB-6548) 是由Akebia Therapeutics和日本田邊三菱公司聯合開發的以吡啶為母核的口服HIF-PHD抑制劑。2019年7月,Akebia與三菱已向日本厚生勞動省共同提交了Vadadustat用于治療CKD貧血癥的新藥上市申請,2020年8月批準上市。
Vadadustat對PHD2的IC50為0.029 μmol/L,對HIF-2α的穩定性優于HIF-1α,血清EPO升高水平隨劑量依懶性增加,在口服約18 小時后血清EPO達到峰值[20],半衰期為4.5 小時[13],耐受性良好,可顯著增加Hb水平,可提高網織紅細胞和總鐵結合的能力,降低血清鐵蛋白和鐵調素水平,沒有觀察到血壓、VEGF、C-反應蛋白和總膽固醇的顯著變化,最常見的不良反應為鼻咽炎、腹瀉、分流管狹窄和便秘等[45-46]。
在名為J01和J03的兩項研究中,評估了Vadadustat和阿法依泊汀對非透析依賴和透析依賴的CKD患者的療效和安全性,304名非透析依賴和323名透析依賴CKD貧血的日本受試者進行了為期52周的治療,Vadadustat治療組Hb水平與對照組阿法依泊汀相比,都能達到預先規定的非劣性標準[47]。
2.2.5 Enarodustat
Enarodustat (JTZ-951) 是Japan Tobacco開發的一種三氮唑并吡啶類口服HIF-PHD抑制劑。Japan Tobacco在日本已完成Enarodustat針對腎性貧血的Ⅲ期臨床試驗,于2020年9月日本獲批上市,我國藥企信立泰獲得該品種的中國開發授權。
Enarodustat對PHD2的IC50為0.22 μmol/L[48],在非透析性CKD貧血患者中的有效性、安全性和維持劑量研究顯示,初次使用ESA (矯正組) 和穩定劑量的ESA (轉化組) 患者被隨機分配,以雙盲方式每天一次接受2 mg、4 mg或6 mg Enarodustat或安慰劑治療6周(1階段),然后進行24周的開放Enarodustat治療,以維持其Hb水平在10.0-12.0 g /dL的目標范圍內 (第2階段)。結果顯示,在矯正組中,每周Hb水平的增加速率呈劑量依賴方式增加。在第1階段中,Enarodustat組和安慰劑組在Hb水平保持在基線的±1.0 g / dL以內,無顯著差異。在第2階段的治療結束時,兩組中超過70%的受試者將Hb水平保持在目標范圍內。校正組和轉化組的平均處方劑量分別為3.58和3.74 mg/天。Enarodustat與鐵調素和鐵蛋白的減少以及總鐵結合能力的增加有關,可以糾正和維持未透析的CKD貧血患者的血紅蛋白水平,并且通常被很好地耐受[49]。
2.2.6 Desidustat
Desidustat是印度Zydus Cadila公司研發的一種新型口服HIF-PHD抑制劑,用于治療接受透析和非透析的CKD貧血患者,Desidustat對PHD2的IC50為11.20 μmol/L,在腎性貧血和炎癥性貧血的大鼠模型中能夠持續性誘導EPO產生[50]。
健康志愿者口服給藥的Ⅰ期臨床研究結果顯示Desidustat耐受性良好,安全有效,血漿EPO平均水平呈劑量依賴性增加,Desidustat的代謝不會因為腎臟病變的嚴重程度而發生變化[51]。II期臨床研究共納入了117例非透析CKD貧血患者,經過6周的治療,與安慰劑相比,在3個Desidustat劑量水平組,主要有效性終點Hb水平的升高均實現了有統計學意義的顯著差異,次要終點Hb反應率在3個Desidustat劑量水平均超過60%。安全性方面,沒有觀察到嚴重不良事件,生命體征、心電圖參數或安全化驗值均無明顯變化[52]。
Zydus已啟動了兩項Desidustat III期臨床試驗,計劃納入未接受透析的CKD患者和正在接受透析的CKD患者。
2.2.7 Molidustat
Molidustat (BAY-85-3934) 是由Bayer公司研發的口服HIF-PHD抑制劑。Molidustat原本是Bayer的心臟病藥物,目前已完成慢性腎病或終末期腎病相關貧血Ⅱ期臨床試驗,有望進入Ⅲ期臨床。
Molidustat可以抑制3種PHD亞型 (IC50 PHD1: 0.48μM; PHD2: 0.28μM; PHD3: 0.450 μM)[45]。在腎性貧血小鼠模型中,Molidustat除糾正貧血以外,還有降血壓的作用,能使血壓穩定在正常水平;它還能糾正多發性關節炎大鼠模型中炎癥引發的貧血,降低單核細胞趨化蛋白-1和鐵調素mRNA的表達[12,53]Molidustat平均半衰期是7~13小時[54],其主要代謝產物為無藥理活性的N-葡萄糖醛酸苷[55]。在三項共計納入400多例CKD患者為期16周的隨機IIb期劑量學研究中,Molidustat與安慰劑相比,可糾正Hb水平或維持在與那些繼續接受ESAs治療的患者相當的水平,副作用可控[56]。
2.2.8 其他HIF-PHD抑制劑研究進展
恒瑞醫藥于2019年1月28日偕同同蘇州市盛迪亞生物技術和中國藥科大學向國家食藥監局提交DDO-3055片臨床研究申請辦理獲審理;2018年11月,東陽光藥HEC53856被默認許可臨床試驗,此外,三生制藥的產品正在進行臨床審評審批。
3 HIF-PHD抑制劑的潛在風險
HIF-PHD抑制劑最常見的不良反應是惡心、腹瀉等胃腸道反應,Hb水平過分增高也是其使用的一個潛在風險[57],HIF-PHD抑制劑的致癌風險也是未來需要關注的重點,其可誘導VEGF基因的轉錄和表達,從而促進血管形成、提高了腫瘤細胞的供給、幫助其存活,甚至通過血管通透性增加了腫瘤轉移的風險[58],此外,VEGF還與眼內新生血管疾病有關,包括年齡相關性黃斑變性、脈絡膜新生血管和糖尿病視網膜病變[59]。由于HIF-PHD抑制劑能刺激紅細胞的生物合成,提高個體氧的運輸能力,因此很可能會被濫用于體育運動中[30]。
4 結語
HIF-PHD抑制劑是一類通過模擬機體在高海拔地區或缺氧條件下,靶向抑制PHD來穩定HIF,進而刺激內源性EPO產生且維持其正常生理水平,上調轉鐵蛋白受體表達、增加鐵吸收、促進紅細胞成熟的新型治療貧血適應癥的藥物。相比注射ESAs和輸血,HIF-PHD抑制劑口服的給藥方式避免了前者注射或靜滴給藥的不便和冷藏要求,對正在接受透析和未透析的CKD貧血患者更加受益。雖然就目前研究來看,HIF-PHD抑制劑優點突出,但對其潛在的安全性問題不容忽視。未來臨床研究,仍需長期評估傳統ESAs和新型HIF-PHD抑制劑的治療效果,評估HIF-PHD抑制劑對腫瘤、心血管事件、高血壓和骨質疏松等潛在風險。另外,已上市和在研藥物的臨床研究顯示,目前的HIF-PHD抑制劑缺乏對PHD亞型的特異性選擇,因此,開發特異性選擇的HIF-PHD抑制劑是未來研究工作的重點。
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
