聚丙烯酸螯合樹脂的制備及其吸附堿性溶液中鈾的性能
2021-06-02 13:09:00陳樹森郎哲思
濕法冶金 2021年3期
封 宇,陳樹森 ,任 宇,郎哲思
(1.核工業北京化工冶金研究院,北京 101149;2.中國鈾業有限公司,北京 100013)
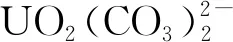
與離子交換樹脂相比,螯合樹脂對重金屬離子的結合能力更強,選擇性更高,具有較好的吸附性能,在金屬離子的富集、分離、分析和回收等領域被廣泛應用[6-7]。如果能制備一種既含有強堿基團,又含有螯合基團的樹脂,利用雙功能基團的協同吸附作用,將有望提高樹脂在高濃度碳酸鹽環境中的鈾吸附容量。
多胺型螯合樹脂對過渡金屬和重金屬離子具有較好的選擇性和很高的吸附容量,對金、銀和鎳等重金屬離子的吸附得到了廣泛關注,目前已在重金屬廢水處理領域得到應用[7-10]。試驗制備了一種多胺型螯合樹脂并將其與碘甲烷進行季銨化反應,得到含強堿基團的螯合樹脂,并考察其在高濃度碳酸鹽溶液中對鈾的吸附能力。
1 試驗部分
1.1 儀器與試劑
主要儀器:TENSONII型紅外光譜儀(美國Bruker公司),GEMINI V2380比表面分析儀(美國麥克公司),SHZ-82型氣浴恒溫振蕩器(江蘇金壇市榮華儀器制造有限公司),智能升降恒溫水浴鍋(鞏義市予華儀器有限責任公司),動力機械攪拌器(江蘇金壇市榮華儀器制造有限公司),PHS-25型酸度計(北京精微博科技有限公司),三口瓶、蛇形冷凝管、索氏提取器(北京欣維爾玻璃儀器有限公司)。
主要試劑:U3O8(純度≥99.8%,中國核工業集團公司二七二廠)),過氧化苯甲酰、丙烯酸甲酯、雙甲基丙烯酸乙二醇酯(EGDM)、N,N-二異丙基乙胺(國藥集團化學試劑有限公司),四乙烯五胺(廣東西隴化工有限公司),二乙烯苯(南開大學化工廠,55%),明膠(包頭明膠廠),200#汽油(無錫百施特化工有限公司),甲苯、二甲基甲酰胺、苯乙酮、碘甲烷、濃硫酸、氯化鈉、氫氧化鈉、碳酸鈉、碳酸氫鈉、鹽酸、硫酸鈉、甲醇、丙酮等(分析純),201×7離子交換樹脂(爭光樹脂廠)。
1.2 樹脂的制備
聚丙烯酸螯合樹脂的制備過程如圖1。首先采用懸浮聚合法制備大孔丙烯酸甲酯共聚物(以下簡稱白球);再通過酯基氨解反應在白球上接枝四乙烯五胺,得到螯合樹脂;最后將螯合樹脂與碘甲烷混合使其季銨化得到含有強堿基團的聚丙烯酸螯合樹脂。
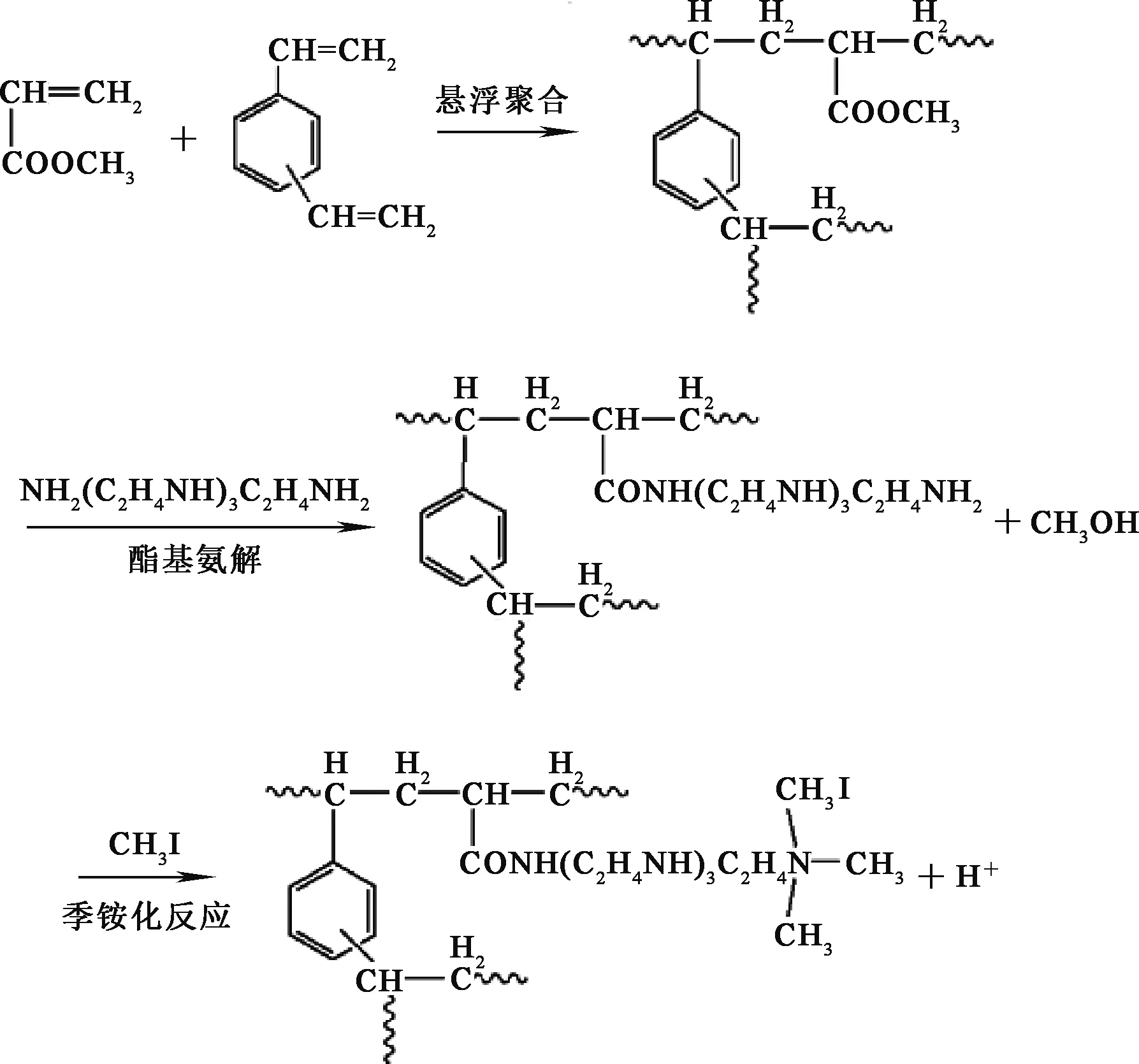
圖1 聚丙烯酸螯合樹脂的制備過程
大孔丙烯酸甲酯共聚物(白球)的制備[11]:在裝有攪拌器、溫度計、冷凝管的500 mL三口燒瓶中加入150 mL去離子水、1.5 g明膠和22.5 g氯化鈉,升溫至45 ℃,待明膠和氯化鈉全部溶解,得到水相。在燒杯中依次加入2.4 g引發劑過氧化苯甲酰、22 g丙烯酸甲酯、2.4 g交聯劑二乙烯苯(或與交聯劑EGDM按質量比4/1混合),8.5 mL致孔劑200#汽油。待引發劑全部溶解,將其加入到上述水相中,通過調節攪拌速度控制液滴粒度,升溫至75 ℃保溫3 h,再升溫至85 ℃保溫10 h后停止反應,得到白球。所得白球經熱水洗滌、丙酮抽提,在60 ℃下烘干后篩分,備用。
螯合樹脂的制備(白球酯基氨解):上一步所得白球在甲苯中溶脹8 h后,加入過量四乙烯五胺,升溫至120 ℃反應24 h,氨解反應后得到螯合樹脂。將樹脂用自來水洗滌至中性,再以2 mol/L鹽酸溶液浸泡攪拌2 h,用去離子水洗滌至中性,用濃度為2 mol/L NaOH溶液浸泡攪拌2 h,再以去離子水洗滌至中性,丙酮抽提,真空干燥后備用。
聚丙烯酸螯合樹脂的制備(螯合樹脂季銨化):將上述螯合樹脂與過量碘甲烷混合使發生季銨化反應,得到聚丙烯酸螯合樹脂(含強堿基團的螯合樹脂)。螯合樹脂用體積分數50%的甲醇溶液溶脹3 h,再與碘甲烷及縛酸劑N,N-二異丙基乙胺混合,升溫至45 ℃后保溫75 h,用自來水洗滌至中性,以2 mol/L鹽酸溶液浸泡攪拌2 h,用去離子水洗滌至中性,再用2 mol/L的NaOH溶液浸泡攪拌2 h,以去離子水洗滌至中性,得到產物聚丙烯酸螯合樹脂。
對烘干后的聚丙烯酸螯合樹脂用紅外光譜進行結構表征。樹脂交換容量參照國標GB/T 5760—2000進行測定。
1.3 樹脂對鈾的吸附
稱取0.2 g聚丙烯酸螯合樹脂于具塞三角瓶中,加入堿性含鈾溶液200 mL,蓋緊塞子置于25 ℃空氣浴中進行靜態吸附。吸附平衡后,測定尾液中鈾質量濃度,計算樹脂對鈾的吸附容量。
吸附動力學:含鈾溶液中,鈾酰離子、碳酸根、碳酸氫根質量濃度分別為0.3、36、14 g/L;溶液100 mL與樹脂0.1 g混合振蕩0.5~15 h,然后分析溶液中鈾質量濃度,計算樹脂對鈾的吸附量,繪制吸附動力學曲線,再以動力學模型對數據進行擬合。
吸附等溫線:含鈾溶液中,鈾酰離子、碳酸根、碳酸氫根質量濃度分別為0.05~0.8、36、14 g/L;溶液100 mL與樹脂0.1 g混合后振蕩6 h,然后測定樹脂對鈾的吸附量,繪制吸附等溫線,再以吸附等溫模型對數據進行擬合。
2 試驗結果與討論
2.1 樹脂的制備
2.1.1 懸浮聚合反應中交聯劑對成球的影響
首先,采用懸浮聚合法制備丙烯酸甲酯-二乙烯苯白球[11],在對白球進行酯基氨解反應制備螯合樹脂過程中發現,螯合樹脂的機械強度較差,樹脂表面有裂痕,出現碎裂現象。這是因為二乙烯苯的聚合活性比丙烯酸甲酯的大[6,12],反應速率更快,聚合初期交聯度大,后期交聯度迅速下降,所形成的共聚物交聯結構不夠均勻,導致樹脂機械強度較差。
在體系中加入適量聚合活性相對較小的交聯劑EGDM,考察混合交聯劑對白球機械強度的影響,試驗結果見表1。當交聯度(交聯劑占交聯劑與丙烯酸甲酯總質量的百分比)>10、二乙烯苯與EGDM質量比為4/1時,經酯基氨解反應后白球仍保持顆粒完整,無碎裂現象,表明混合交聯劑與丙烯酸甲酯形成的共聚物結構更均勻、機械強度更好;隨交聯劑用量繼續增加,雖然骨架結構趨于緊密,但白球上可以引入酯基的功能基團的數量會減少,導致樹脂弱堿基團交換容量下降。

表1 交聯劑用量對螯合樹脂交換容量的影響
2.1.2 白球的酯基氨解
對所得白球進行酯基氨解反應過程中,四乙烯五胺分子兩端的伯胺基團均可與丙烯酸甲酯進行反應,四乙烯五胺用量不足會導致其與丙烯酸甲酯以2/1的比例進行反應,使下一步季銨化反應可引入的強堿基團的反應位點減少。四乙烯五胺用量對螯合樹脂弱堿基團交換容量的影響試驗結果見表2。
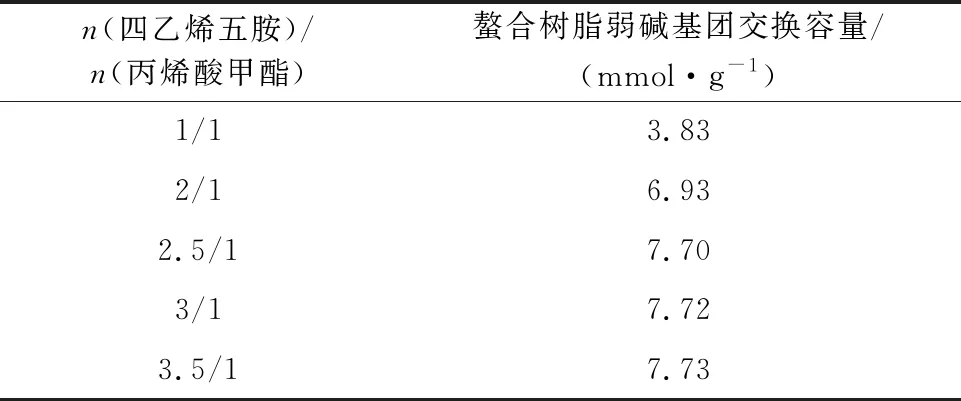
表2 四乙烯五胺用量對螯合樹脂弱堿基團交換容量的影響
由表2看出:體系中,四乙烯五胺與丙烯酸甲酯的物質的量比為1/1時,所得螯合樹脂弱堿基團的交換容量僅為3.83 mmol/g;而當四乙烯五胺與丙烯酸甲酯的物質的量比為2.5/1時,樹脂弱堿基團交換容量提高到7.70 mmol/g。
在交聯劑二乙烯苯與EGDM質量比為4/1、交聯度10%、四乙烯五胺與丙烯酸甲酯物質的量比為2.5/1、反應時間24 h條件下,反應溫度對樹脂弱堿基團交換容量的影響試驗結果如圖2所示。
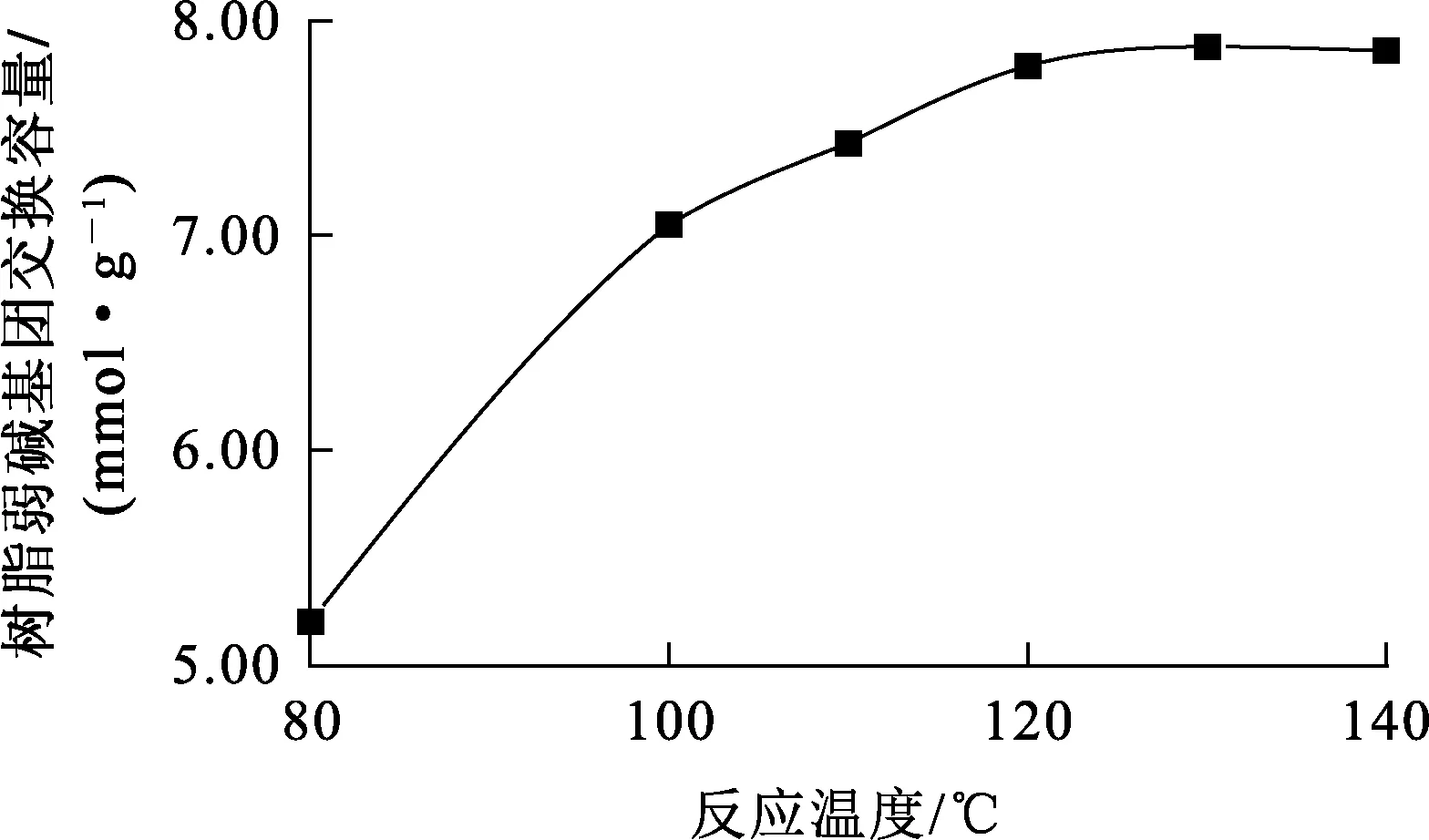
圖2 反應溫度對樹脂弱堿基團交換容量的影響
由圖2看出:酯基氨解反應最佳溫度為130 ℃, 此條件下,樹脂弱堿基團交換容量為7.9 mmol/g;繼續升高反應溫度,樹脂弱堿基團交換容量提高不明顯。
在交聯劑二乙烯苯與EGDM質量比為4/1、交聯度為10%、四乙烯五胺與丙烯酸甲酯物質的量比為2.5/1、反應溫度130 ℃條件下,反應時間對樹脂弱堿基團交換容量的影響試驗結果如圖3所示。
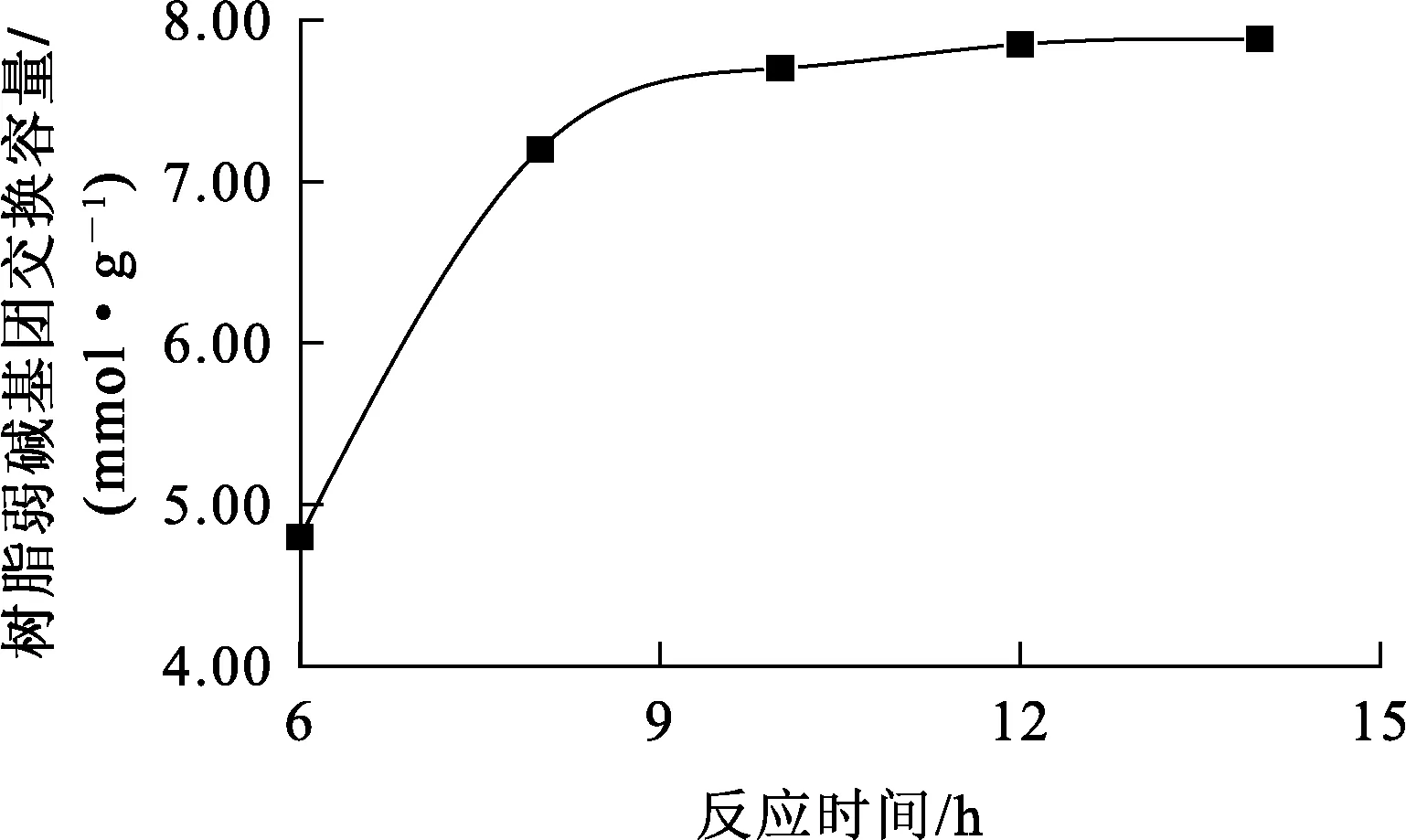
圖3 反應時間對樹脂弱堿基團交換容量的影響
由圖3看出:酯基氨解反應最佳時間為12 h,此條件下,樹脂弱堿基團交換容量為7.9 mmol/g;繼續延長反應時間,樹脂弱堿基團交換容量提高不明顯。
2.1.3 螯合樹脂的季銨化
用碘甲烷對所得螯合樹脂進行季銨化引入弱堿基團,以獲得聚丙烯酸螯合樹脂以提高樹脂對鈾的吸附性能。由于季銨化反應過程中會產生H+,而中和產生的H+有利于反應正向進行。因此,選擇有機堿性物質N,N-二異丙基乙胺作縛酸劑中和H+。此縛酸劑有較大位阻,不與碘甲烷反應,可減少副反應發生。
在碘甲烷與螯合樹脂物質的量比為5/3、縛酸劑和螯合樹脂的物質的量比為5/1、反應時間75 h條件下,反應溫度對樹脂強堿基團交換容量的影響試驗結果見表3。可以看出:隨反應溫度升高,樹脂強堿基團交換容量提高;反應溫度達45 ℃時,樹脂強堿基團交換容量最大,為3.90 mmol/g。
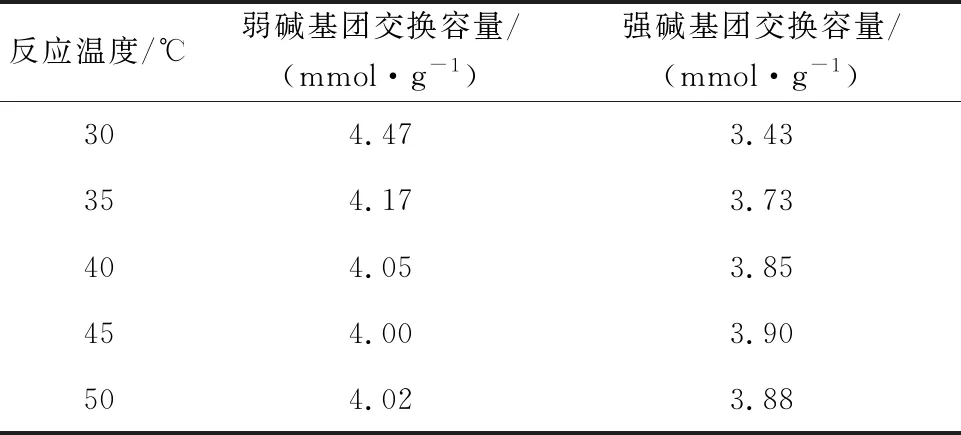
表3 反應溫度對季銨化后樹脂交換容量的影響
在碘甲烷與螯合樹脂的物質的量比為5/3、縛酸劑和螯合樹脂的物質的量比為5/1、反應溫度45 ℃ 條件下,反應時間對樹脂強堿基團交換容量的影響試驗結果見表4。可以看出:隨反應進行,樹脂強堿基團交換量提高;反應時間為70 h時,樹脂強堿基團交換量最大,為3.96 mmol/g。
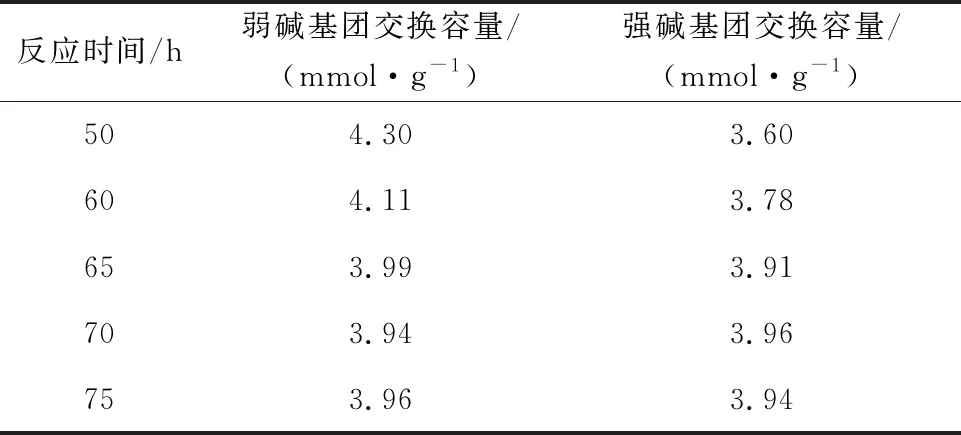
表4 反應時間對季銨化后樹脂交換容量的影響
在縛酸劑和螯合樹脂的物質的量比為5/1、反應溫度45 ℃、反應時間70 h條件下,碘甲烷用量對樹脂強堿基團交換容量的影響試驗結果見表5。
對照組發生皮下出血、惡心、大便潛血陽性的例數分別為3例、5例及3例,不良反應總發生率為24.44%(11/45);觀察組發生皮下出血、大便潛血陽性例數分別為1例、1例,不良反應總發生率為4.44%(2/45)。觀察組的不良反應發生率明顯低于對照組,差異具有統計學意義(P<0.05),見表3。
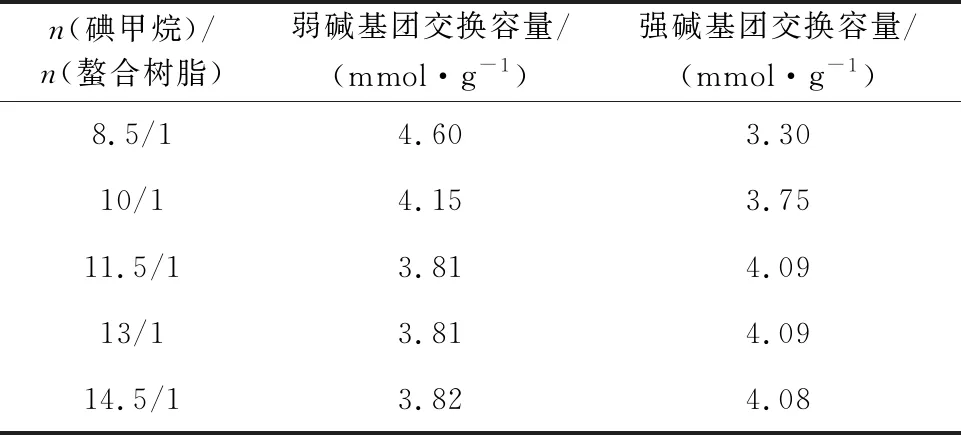
表5 碘甲烷用量對季銨化后樹脂交換容量的影響
由表5看出:隨碘甲烷與螯合樹脂的物質的量比增加,樹脂強堿基團交換容量增大;碘甲烷與螯合樹脂的物質的量比達11.5/1時,樹脂強堿基團交換容量達最大,為4.09 mmol/g。
在碘甲烷與螯合樹脂的物質的量比為11.5/1、反應時間70 h、反應溫度45 ℃條件下,縛酸劑用量對樹脂強堿基團交換容量的影響試驗結果見表6。可以看出:隨縛酸劑與螯合樹脂的物質的量比增大,樹脂強堿基團交換容量增大;縛酸劑與螯合樹脂的物質的量比為7/1時,樹脂強堿基團交換容量最大,為4.2 mmol/g。
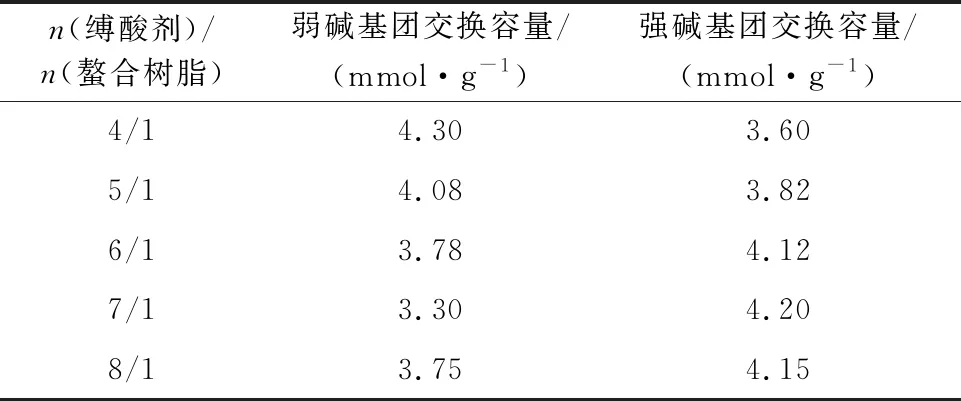
表6 縛酸劑用量對樹脂交換容量的影響
2.1.4 樹脂的紅外光譜分析
圖4曲線a為大孔丙烯酸甲酯共聚物(白球)的特征譜線,1 736 cm-1處為酯羰基吸收峰;圖4曲線b為螯合樹脂的特征譜線,1 736 cm-1處的酯羰基吸收峰明顯減弱,1 649、1 560 cm-1處出現了酰胺Ⅰ和酰胺Ⅱ吸收峰,說明酯基氨解反應成功引入了螯合基團;圖4曲線c為含強堿基團的螯合樹脂(聚丙烯酸螯合樹脂)的特征譜線,1 560 cm-1處的吸收峰偏移至1 600 cm-1處,且1 351 cm-1處出現了C—N伸縮振動峰[13],說明季銨化反應有效地在螯合樹脂上引入了強堿基團。
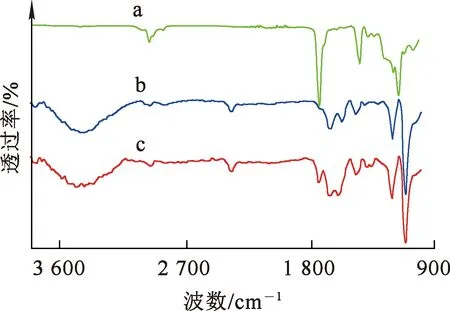
a—大孔丙烯酸甲酯共聚物;b—螯合樹脂;c—含強堿基團的螯合樹脂。圖4 樹脂的紅外光譜
2.2 樹脂對鈾的吸附
2.2.1 樹脂對鈾的飽和吸附量
選擇堿性浸出液對上述樹脂進行靜態吸附,其中鈾、碳酸根、碳酸氫根質量濃度分別為0.8、36、14 g/L。吸附達到平衡后,更換溶液繼續進行吸附,當吸附尾液中鈾質量濃度等于鈾初始質量濃度時,樹脂達到吸附飽和,分析樹脂的飽和吸附容量,結果見表7。在上述鈾溶液中,聚丙烯酸螯合樹脂在3次吸附后達到飽和,對鈾的飽和吸附容量為126.6 mg/g。

表7 樹脂飽和吸附容量
配制pH=11、鈾質量濃度0.8 g/L、碳酸氫根質量濃度14 g/L、不同碳酸根質量濃度的堿性溶液,考察碳酸根質量濃度對聚丙烯酸螯合樹脂和強堿性樹脂201×7吸附鈾的影響,試驗結果如圖5所示。可以看出:2種樹脂對鈾的吸附容量均隨碳酸根質量濃度增大而顯著下降,但聚丙烯酸螯合樹脂的吸附下降趨勢比201×7的吸附下降趨勢要小。碳酸根會與三碳酸鈾酰配陰離子發生競爭吸附,導致吸附容量下降。
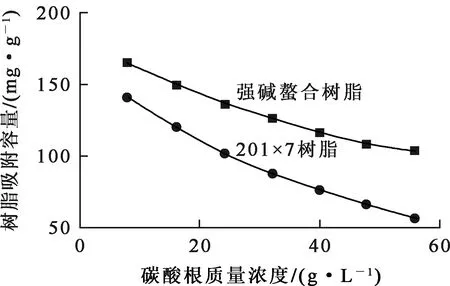
圖5 碳酸根質量濃度對樹脂吸附鈾的影響
2.2.3 吸附動力學
繪制聚丙烯酸螯合樹脂對鈾的吸附量隨吸附時間變化的動力學曲線,然后根據曲線以準一級動力學模型及準一級動力學模型[14-16]對數據進行擬合。
(1)
(2)
式中:qe—吸附平衡時樹脂對鈾的吸附容量,mmol/g;qt—吸附時間t時樹脂對鈾的吸附容量,mmol/g;k1—準一級模型的吸附速率平衡常數,min-1;k2—準二級模型的吸附速率平衡常數,g/(mg·min)。 吸附動力學曲線及擬合曲線如圖6所示。
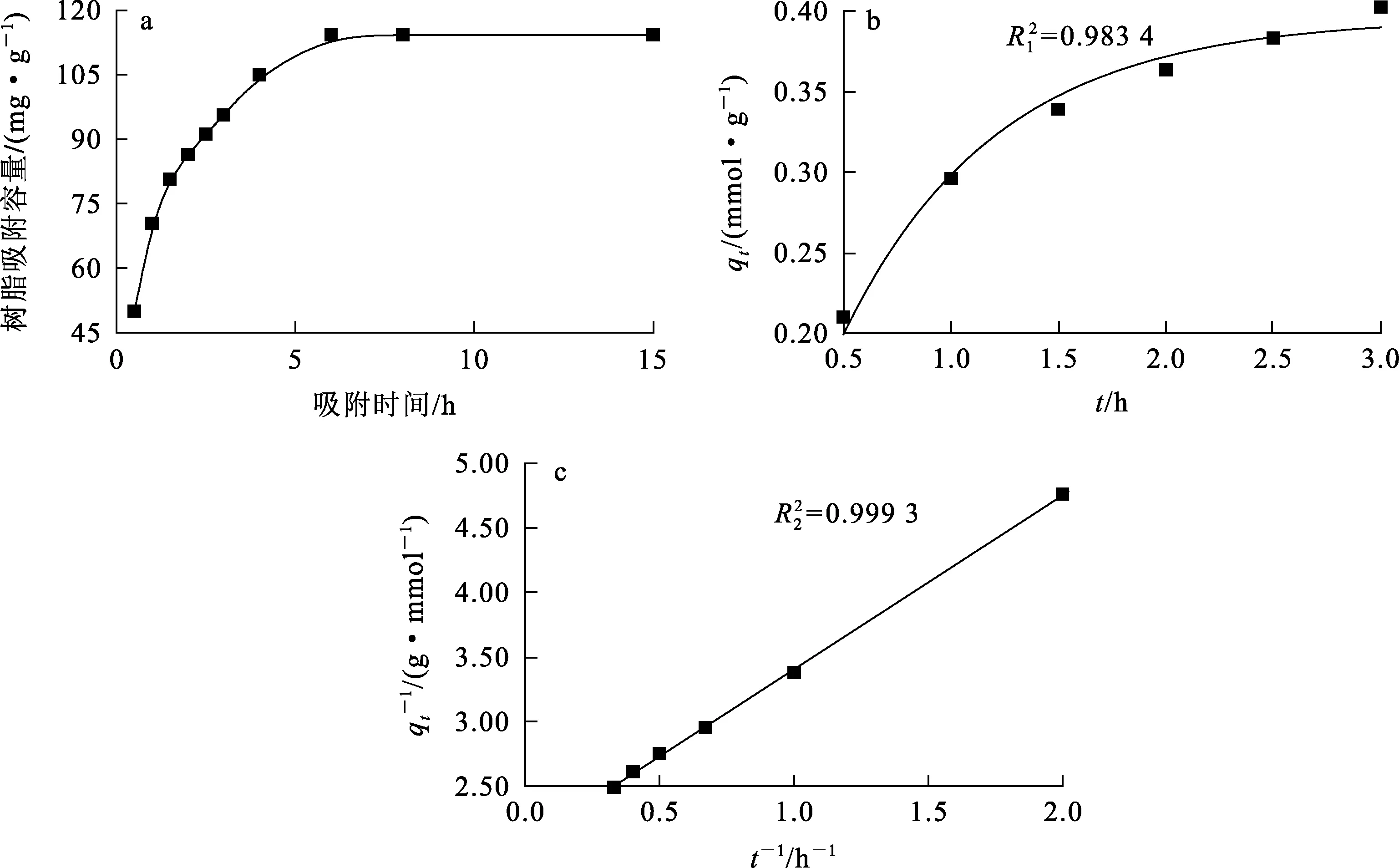
a—吸附動力學曲線;b—準一級動力學擬合曲線;c—準二級動力學擬合曲線。圖6 聚丙烯酸螯合樹脂對鈾的吸附動力學曲線及擬合曲線
2.2.4 吸附等溫線
以吸附尾液中鈾質量濃度為橫坐標,樹脂對鈾的吸附量為縱坐標繪制吸附等溫線,然后以Langmuir及Freundlich等溫吸附模型[14-16]對數據進行擬合。
Langmuir吸附等溫模型,
(3)
Freundlich吸附等溫模型
(4)
式中:ce—吸附平衡時溶液中鈾濃度,mmol/L;qe—吸附平衡時樹脂對鈾的吸附容量,mmol/g;qm—樹脂對鈾的飽和吸附容量,mmol/g;kL—Langmuir模型吸附平衡常數,L/mg;kF—Freundlich模型吸附平衡常數,mg/(g·mg1/n);n—無因次參數。吸附等溫線及擬合曲線如圖7所示。
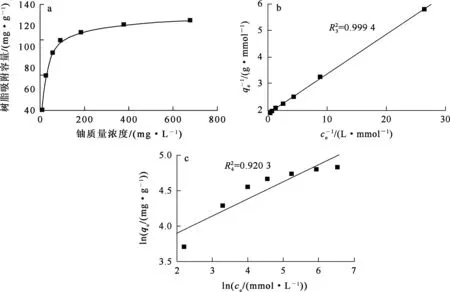
a—吸附等溫線;b—Langmuir等溫吸附擬合曲線;c—Freundlich等溫吸附擬合曲線。圖7 聚丙烯酸螯合樹脂對鈾的吸附等溫線及擬合曲線
3 結論
采用懸浮聚合法,以二乙烯苯和EGDM混合溶液作交聯劑制備大孔丙烯酸甲酯共聚物,經酯基氨解、季銨化反應,可以制得機械強度較高的聚丙烯酸螯合樹脂。在含有高濃度碳酸根/碳酸氫根溶液中,所制備樹脂對鈾有較好的選擇吸附性,吸附過程受化學反應控制,屬于單層吸附。鈾、碳酸根、碳酸氫根質量濃度分別為0.8、36、14 g/L條件下,該螯合樹脂對鈾的飽和吸附容量可達126.6 mg/g。
