ZrO2改性對Ni/SBA-15催化二苯并呋喃加氫脫氧的促進作用研究
2021-06-02 14:01:36杜朕屹李文英
燃料化學學報 2021年5期
關鍵詞:催化劑
郭 良,劉 迪,杜朕屹,馮 杰,李文英
(太原理工大學省部共建煤基能源清潔高效利用國家重點實驗室,山西太原030024)
熱解技術是低階煤分級分質利用的核心,煤焦油是熱解技術的主要產物之一。然而,中低溫煤焦油中存在大量的含氧化合物[1],這些含氧化合物降低了油品熱值和穩定性,降低了油品品質,因此,必須對中低溫煤焦油進行加氫精制以滿足其作為燃料應用的要求[2]。中低溫煤焦油中氧原子主要以酚類、呋喃類形式存在,醚、酮、羧酸、醇、酯等物質的含量相對較低[3,4]。同時各含氧化合物的加氫活性不同,相同反應條件下二苯并呋喃反應活性最低[5,6]。因而以中低溫煤焦油中含量高、分子量大、反應活性低的二苯并呋喃為模型化合物可以反映催化劑的加氫脫氧性能[7?9],所得結果具有代表性。Wang等[10]研究了DBF在Pt/介孔ZSM-5分子篩、Pt/微孔ZSM-5分子篩以及Pt/γ-Al2O3催化劑上的加氫脫氧反應,發現高空速下DBF在Pt/介孔ZSM-5催化劑上脫氧程度更高,這是因為介孔ZSM-5載體有規整的介孔孔道,有利于反應物和產物的傳質,同時ZSM-5載體表面較強的酸性位點也有利于脫水反應的發生,有利于含氧化合物中氧的脫除。Wang等[11]研究了DBF在SBA-15負載的貴金屬催化劑上的加氫脫氧反應,發現貴金屬Pt具有較好的加氫活性,Ru具有較高的脫氧產物選擇性。然而,貴金屬催化劑的成本相對較高,而過渡金屬Ni價格低廉,適合替代貴金屬催化劑進行工業化應用。加氫脫氧反應中過渡金屬Ni有較高的加氫活性,但其斷裂C–O鍵的能力相對較弱,為提高Ni基催化劑性能,研究者已展開如下研究:第一,結合其他活性金屬的性質及作用,制備雙金屬催化劑。Dong等[12]研究了DBF在Ni、Pt單金屬催化劑與NiPt雙金屬催化劑上的加氫脫氧反應,發現雙金屬催化劑的活性更高,這是因為雙金屬催化劑上Ni、Pt的協同作用可以促進C–O鍵的斷裂,有利于反應的進行。第二,利用可還原金屬氧化物在還原過程中部分還原產生的氧空位,來促進含氧化合物中C–O鍵的活化、斷裂。文獻[13]研究了苯酚在不同載體負載的Pd催化劑上的加氫脫氧反應,發現Pd/TiO2、Pd/ZrO2催化劑活性較好,反應結束時產物苯的選擇性較高,這是因為TiO2、ZrO2載體對含氧化合物中氧原子有較強的吸附作用,從而促進了含氧化合物中氧的脫除,提高了脫氧產物的選擇性。Zhao等[14]研究了順丁烯二酸酐在Ni/ZrO2催化劑上的加氫脫氧反應,發現相對缺電子的氧空位可以吸附含氧化合物中帶有孤電子的氧原子,從而活化并削弱C=O鍵,結合活性金屬Ni良好的加氫能力,實現C=O的斷裂,其中,吸附在氧空位上的氧原子與Ni上解離的氫結合并以水的形式脫除。
結合當前加氫脫氧反應中Ni基催化劑的研究進展,本研究以DBF為模型化合物,選用惰性、擴散阻力小的有序介孔SBA-15為載體,過渡金屬Ni為活性金屬,通過等體積浸漬方式制備了不同ZrO2含量的Ni/Zr-SBA-15催化劑及Ni/ZrO2催化劑,探究了ZrO2改性對Ni/SBA-15催化劑結構特性與反應活性的影響。
1 實驗部分
1.1 催化劑的制備
實驗中所用的SBA-15載體購于江蘇先豐納米材料科技有限公司。Ni/Zr-SBA-15催化劑通過分步等體積浸漬方式得到,具體制備流程為:先將一定量SBA-15載體與相應質量硝酸氧鋯水溶液混合,室溫攪拌24 h,90℃干燥12 h,之后在馬弗爐中400℃焙燒5 h得到Zr-SBA-15載體。然后以相同步驟將過渡金屬Ni浸漬在Zr-SBA-15載體上得到NiO/Zr-SBA-15催化劑前驅體,將該前驅體在管式爐中經500℃氫氣氣氛下還原4 h得到Ni/Zr-SBA-15催化劑。其中,催化劑上活性金屬Ni的質量分數恒定為5%(質量分數),調變ZrO2在載體中的質量分數為0、2.5%、5%、10%、20%、30%,分 別 記 作Ni/SBA-15、Ni/2.5Zr-SBA-15、Ni/5Zr-SBA-15、Ni/10Zr-SBA-15、Ni/20Zr-SBA-15、Ni/30Zr-SBA-15催化劑。此外,以ZrO2為載體制備了Ni/ZrO2催化劑作為對照,其中,ZrO2載體是將硝酸氧鋯在馬弗爐中500℃焙燒5 h得到,之后以等體積浸漬方式將活性金屬Ni負載在ZrO2載體上得到Ni/ZrO2催化劑,制備流程與Ni/Zr-SBA-15催化劑相同。
1.2 催化劑的表征
采用日本Rigaku Ultima IV型X射線衍射儀(X-ray Diffraction,XRD),以Cu靶為輻射源(λ=0.154 nm),通過小角XRD測定載體的有序結構,0.6°?5°掃描,掃描速率為0.5(°)/min;通過大角XRD測定催化劑的晶相結構,10°?80°掃描,掃描速率為4(°)/min。
采用美國Quantachrome Autosor-iQ物理吸附儀,通過Multi point BET方法分析樣品的等溫吸附-脫附曲線,得到樣品的比表面積;通過分析吸附等溫線相對壓力在0.99(p/p0=0.99)下的N2吸附數據得到樣品的孔容;通過對脫附支進行BJH模型分析得到樣品的孔徑分布。測試前將樣品在300℃真空條件下預處理3 h以脫除樣品表面吸附的水分及雜質,之后在?196℃下進行N2吸附-脫附實驗。
采用美國麥克Autochem II-2920化學吸附儀,通過氫氣程序升溫還原(H2-temperature programmed reduction,H2-TPR)分析催化劑的還原特性。具體操作流程為:將0.02 g焙燒后的催化劑前驅體置于U-形石英樣品管中,在Ar氣氛下400°C預處理1 h,以除去樣品表面吸附的水及雜質,待樣品溫度降至100°C時將載氣切換為體積分數為10%的H2/Ar氣氛,并以10 °C/min的升溫速率升至900 °C進行程序升溫還原實驗,過程中以熱導檢測器(TCD)記錄信號,得到H2-TPR譜圖。
采用上述Autochem II-2920化學吸附儀,通過氫氣程序升溫脫附(H2-temperature programmed desorption,H2-TPD)分析催化劑上活性位點的數目。具體操作流程為:將0.1 g催化劑置于U-形石英樣品管中,在體積分數為10%的H2/Ar氣氛下500°C預處理2 h,Ar吹掃30 min,待樣品溫度降至50°C時再次以10%的H2/Ar處理2 h,Ar吹掃2 h,之后樣品以10℃/min升溫速率升至400℃進行氫氣程序升溫脫附實驗,脫附過程中TCD檢測記錄信號,得到H2-TPD譜圖。同時根據催化劑H2-TPD分析結果計算了催化劑上活性金屬Ni的分散度,計算公式如(1)所示,其中,nH2為H2-TPD中脫附氫氣物質的量,nNi為催化劑中活性金屬Ni物質的量,SF為化學計量因子(Ni/H),SF= 2。

采用上述Autochem II-2920化學吸附儀,通過氧脈沖分析催化劑上氧空位的相對含量。具體操作流程如下:將0.02 g催化劑置于U-形石英樣品管中,在體積分數為10%的H2/Ar氣氛下500℃預處理2 h,He吹掃30 min并開始降溫,待樣品溫度降至340℃時切換為體積分數3%的O2/He氣氛進行氧脈沖實驗,脈沖間隔為3 min,通過TCD檢測記錄信號,直至脈沖信號峰強度不發生變化時結束實驗。通過計算催化劑在脈沖過程中氧氣的吸附量,得到催化劑中氧空位的相對含量。
采用吡啶原位吸附紅外光譜測定催化劑上Br?nsted、Lewis酸量,設置紅外光譜儀(德國Bruker,TENSOR 27)分 辨 率 為4 cm?1,譜 圖 累 加32次,譜圖采集為600–4000 cm?1。其中,1540 cm?1處吸收峰為Br?nsted酸位點特征峰,1450 cm?1處吸收峰為Lewis酸位點特征峰。同時測定了不同脫附溫度下酸性位點的特征峰,以反映催化劑酸強度的變化,其中,脫附溫度為150℃時,得到催化劑的總酸量,脫附溫度為300℃時,得到催化劑上中強酸酸量。
采用日本島津AXIS Supra的X射線光電子能譜(X-ray photoelectron spectroscopy,XPS),得到催化劑中氧空位及表面原子的相關信息。XPS的激發源為單色Al靶,在譜圖分析前以污染碳C 1s峰結合能284.8 eV對譜圖進行荷電校正。
1.3 催化劑性能評價
在高壓反應釜中對催化劑加氫脫氧性能進行評價。反應原料由3.0%的DBF、1.0%正十二烷和96.0%正癸烷組成,催化劑質量為60 mg,反應溫度為280℃,室溫下初始氫氣壓力為4 MPa,加熱至280℃時氫氣壓力升至6.5 MPa,反應180 min,過程中每10 min從取樣管在線取樣,并通過氣相色譜-質譜-氫火焰離子化檢測器聯用儀(GC-MSFID)對產物組成進行定性(MS)和定量(FID),色譜柱為HP-5ms(30 m×0.25 mm×0.25μm)。以正十二烷為內標通過內標法對產物組成進行定量,并計算反應過程中催化劑上DBF轉化率(xDBF)、產物選擇性(si)、產物收率(yi)、碳平衡、DBF反應速率(rDBF)及目標產物BCHs生成速率(rBCHs),具體計算公式如下所示:

式中,n0為反應初始時DBF物質的量(mol);n為 反應過程中DBF物質的量(mol);ni為反應過程中某產物i的物質的量(mol);t為反應時間(min);mcat為催化劑質量(g)。為了對比催化劑本征反應活性,在低轉化率下計算了DBF本征反應速率、目標產物BCHs,包含聯環己烷(Bicyclohexyl,BCH)及其同分異構體環戊基甲基環己烷(Cyclopentylmethylcyclohexane,CPMCH)的生成速率及產物選擇性。在計算DBF反應速率時t取10 min,在計算BCHs生成速率及產物選擇性時t取20 min。
2 結果與討論
2.1 催化劑的表征
2.1.1 有序介孔結構及晶相結構
圖1 為SBA-15與ZrO2改性SBA-15的小角XRD譜圖。由圖1可以看到,SBA-15、2.5Zr-SBA-15、5Zr-SBA-15載體在2θ為0.8°、1.4°、1.6°處有明顯的衍射峰,分別對應載體的(100)、(110)、(200)特征峰,表明這些載體均有規整的孔道,且孔道符合二維六方結構[15],同時也說明適量ZrO2的添加(ZrO2含量低于5%,質量分數)仍然可以使SBA-15載體的有序介孔結構得以保持,而當ZrO2含量大于5%時,可以看到載體的小角XRD衍射峰逐漸減弱并消失,表明過量ZrO2的添加填充到了SBA-15孔道當中,降低了載體的長程有序度[16]。對于ZrO2載體,可以看到其小角XRD譜圖為一條平滑曲線,無衍射峰,表明ZrO2載體沒有有序介孔結構。
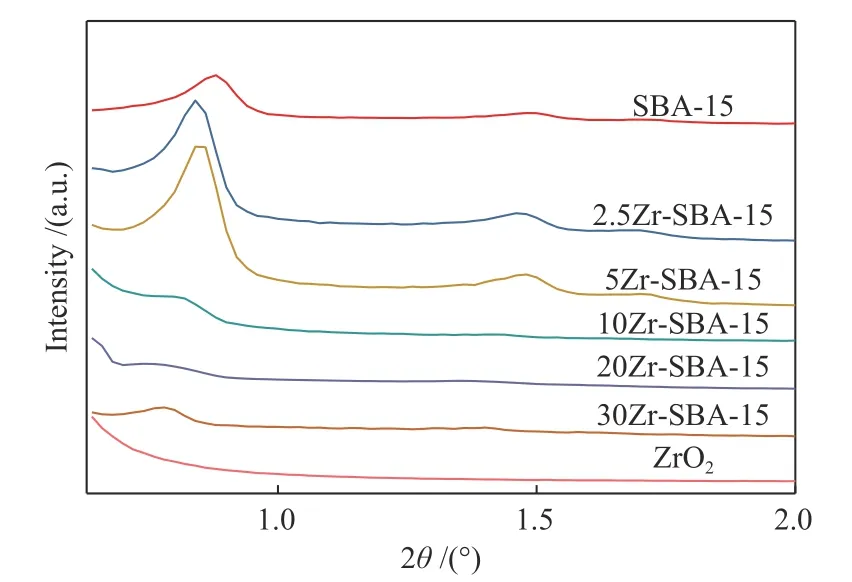
圖1 SBA-15載體與Zr-SBA-15載體的小角XRD譜圖Figure 1 Small-angle XRD patterns of SBA-15 and Zr-SBA-15 supports
圖2 為還原態Ni/Zr-SBA-15催化劑的XRD譜圖,圖2中“t-ZrO2”和“m-ZrO2”分別表示ZrO2的四方相和單斜相。由圖2可以看到,對于Ni/Zr-SBA-15催化劑,其在2θ為44.5°、51.8°和76.4°附近均有衍射峰,分別對應Ni的(111)、(200)和(220)晶面[17];對于未ZrO2改性Ni/SBA-15催化劑,其僅有Ni的衍射峰。隨著ZrO2的添加,催化劑在2θ為30.1°、50.2°和59.7°附近出現衍射峰,為t-ZrO2(四方相),且隨著ZrO2的增加,衍射峰強度增強,這是因為當ZrO2含量較低時(低于10%),ZrO2分散度高,衍射峰強度較低,隨著ZrO2的進一步增加(高于10%),ZrO2開始富集,衍射峰強度增強。對于Ni/ZrO2催化劑,除存在t-ZrO2(四方相)外,其在2θ為24.4°、28.2°和31.4°附近也出現衍射峰,為m-ZrO2(單斜相),且以m-ZrO2為主,因此,Ni/ZrO2催化劑會表現出與Ni/Zr-SBA-15催化劑不同的衍射峰。
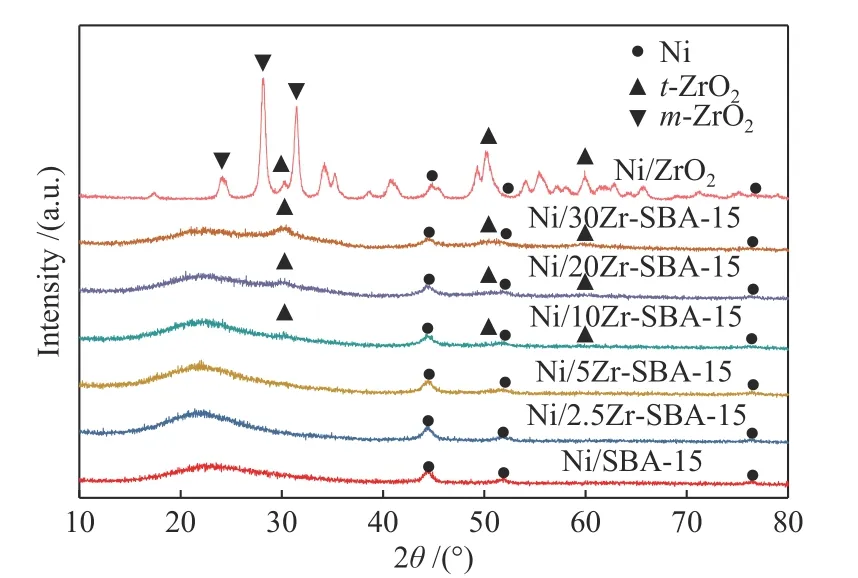
圖2 還原態催化劑的XRD譜圖Figure 2 XRD patterns of reduced catalysts
同時根據謝樂公式,計算了Ni/Zr-SBA-15催化劑上Ni晶粒尺寸,如表1所示。由表1可以看到,Ni/SBA-15催化劑上Ni晶粒尺寸為9.7 nm,隨著ZrO2的增加Ni晶粒尺寸逐漸減小,當ZrO2含量為2.5%、5%、10%、20%、30%時,Ni晶粒尺寸分別為8.2、8.0、7.5、7.3、6.7,表明ZrO2的添加減小了Ni的晶粒尺寸,促進了Ni的分散。對于Ni/ZrO2催化劑,其Ni晶粒尺寸為6.8 nm,與Ni/30Zr-SBA-15催化劑晶粒尺寸相近。

表1 催化劑的化學性質Table 1 Chemical properties of catalysts
2.1.2 比表面積及孔徑分布
圖3 (a)、(b)分別為Ni/Zr-SBA-15催化劑的氮氣物理吸附-脫附曲線及孔徑分布。可以看到,該催化劑的氮氣物理吸附-脫附等溫線為IV型吸附-脫附等溫線,且有明顯的H1型滯后環,表明該催化劑為有序介孔結構[18]。與其他催化劑相比,Ni/ZrO2催化劑的吸附-脫附等溫線基本重合,不是IV型吸附-脫附等溫線,無H1型滯后環,表明Ni/ZrO2催化劑沒有有序介孔結構,這與小角XRD的表征結果相吻合。
根據上述催化劑氮氣物理吸附-脫附曲線及孔徑分布圖,表2給出了Ni/Zr-SBA-15催化劑比表面積、孔容及最可幾孔徑。可以看到,Ni/SBA-15催化劑的比表面積為493.6 m2/g,孔容為1.11 cm3/g,最可幾孔徑為7.8 nm。隨著ZrO2的增加,Ni/Zr-SBA-15催化劑的比表面積、孔容逐漸減小,表明ZrO2的添加會堵塞催化劑孔道。當ZrO2添加量為30%時,比表面積降至360.0 m2/g,孔容降至0.77 cm3/g,最可幾孔徑為7.8 nm。同時可以看到,Ni/ZrO2催化劑具有較小的比表面積、孔容,兩者分別為46.9 m2/g和0.13 cm3/g。
2.1.3 催化劑的酸性
圖4 為Ni/Zr-SBA-15催化劑的吡啶吸附紅外光譜譜圖,其中,1450 cm?1處為Lewis酸的特征峰,1540 cm?1處為Br?nsted酸的特征峰[19],可以看到,Ni/SBA-15催化劑在1450 cm?1處吸附峰強度較弱,在1540 cm?1處無吸附峰,表明Ni/SBA-15催化劑有少量的Lewis酸,而沒有Br?nsted酸。與Ni/SBA-15催化劑相比,可以看到,隨著ZrO2的增加,Ni/Zr-SBA-15催化劑在1450 cm?1處吸收峰呈現出先增大后減小的趨勢,1540 cm?1處仍沒有吸收峰,表明ZrO2的添加僅在催化劑中引入了Lewis酸,而沒有Br?nsted酸,且隨著ZrO2的增加Lewis酸量表現出先增加后減少的趨勢。

圖4 催化劑的吡啶紅外光譜譜圖Figure 4 Py-FTIR profiles of catalysts
表3 列出了催化劑中Lewis酸的酸量。由表3可以看到,Ni/SBA-15催化劑的總酸量為8.2 μmol/g,中強酸酸量為3.8μmol/g,催化劑酸性較弱。隨著ZrO2的添加,催化劑酸量增加,這是因為ZrO2中含配位不飽和的Zr3+及Zr4+,其作為Lewis酸位點,有效增強了催化劑酸性。在ZrO2負載量高于10%(質量分數)時,催化劑酸量下降,這是因為過量的ZrO2降低了催化劑比表面積、孔容,從而降低了催化劑酸量。可以看到,Ni/10Zr-SBA-15催化劑的酸量最高,總酸量為84.5μmol/g,中強酸酸量為30.5μmol/g。同時也分析了催化劑中中強酸在總酸量中的占比,可以看到Ni/SBA-5催化劑上該占比為0.46,隨著ZrO2的增加,中強酸在總酸量中的占比不斷減小,當ZrO2添加量為30%時,該占比下降至0.20。以上結果表明,ZrO2改性在催化劑中引入了Lewis酸,且引入的Lewis酸酸性位點以弱酸為主。
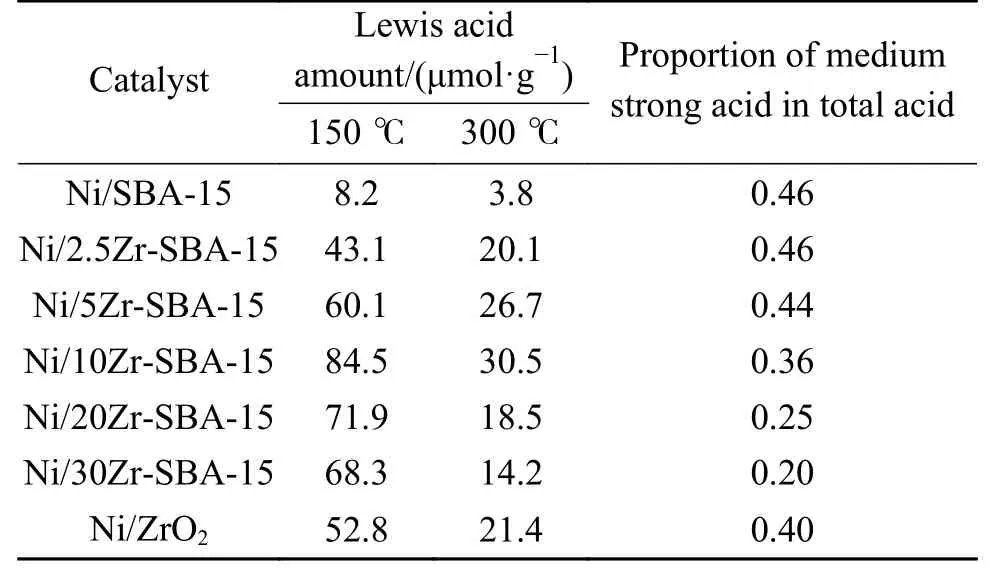
表3 催化劑的酸量Table 3 The acid amount of catalysts
2.1.4 氧空位濃度
圖5 (a)為Ni/Zr-SBA-15催化劑的H2-TPR譜圖。可以看到,ZrO2載體在較高溫度下有氫氣消耗峰,表明ZrO2被部分還原,產生了氧空位。對于Ni/Zr-SBA-15催化劑,可以發現隨著ZrO2的增加,氫氣消耗峰逐漸向高溫方向移動,表明ZrO2的添加增強了Ni物種與載體間相互作用,H2-TPR實驗規律較好。同時結合表1可以發現,ZrO2含量不同催化劑上Ni分散度不同,這也會影響催化劑的還原性能。一方面ZrO2的添加增強了Ni物種與載體間相互作用,提高了Ni物種的還原溫度;另一方面隨著ZrO2的添加,Ni分散度增加,Ni晶粒尺寸減小,Ni物種還原溫度升高,從而也會影響催化劑的還原性能。

圖5 催化劑的H2-TPR (a)及H2-TPD(b)譜圖Figure 5 H2-TPR (a)and H2-TPD(b)profiles of catalysts
同時分析了催化劑在還原過程中氫氣的消耗量,結果如表1所示。可以看到,隨著ZrO2的添加,氫氣消耗量增加,表明在催化劑前驅體的還原過程中,除Ni物種的還原外,還涉及ZrO2的部分還原,且隨著ZrO2的增加,催化劑中氧空位濃度增加。對于Ni/ZrO2催化劑,其H2-TPR譜圖與Ni/Zr-SBA-15催化劑譜圖相比存在明顯差異,根據本文現有數據推測,這是因為Ni/ZrO2催化劑中ZrO2主要以單斜相形式存在,而Ni/Zr-SBA-15催化劑中ZrO2主要以四方相形式存在,四方相ZrO2的存在提高了Ni物種與載體間相互作用,提高了Ni物種的還原溫度,與文獻報道一致[20,21],因此,Ni/ZrO2催化劑與Ni/Zr-SBA-15催化劑表現出不同的還原溫度。
圖5 (b)為Ni/Zr-SBA-15催化劑的H2-TPD譜圖。可以看到,Ni/Zr-SBA-15催化劑在125℃附近有氫氣脫附峰,且隨著ZrO2的添加,脫附峰逐漸變大,表明Ni/Zr-SBA-15催化劑上有更多的氫活性位點。同時表1根據催化劑H2-TPD的表征結果,分析了催化劑上活性金屬Ni的分散度,可以看到隨著ZrO2的增加,H2脫附量增加,催化劑上活性金屬Ni分散度提高,這與XRD上Ni晶粒尺寸的變化趨勢相吻合。而對于Ni/ZrO2催化劑,其活性金屬Ni的分散度僅為6.6%,這是由于ZrO2載體的比表面積較小,不利于活性金屬Ni的分散造成的。
通過O2-Pulse實驗,分析了催化劑中氧空位的相對含量,結果如表1所示。可以看到,Ni/SBA-15催化劑的氧氣消耗量為206.7μmol/g,由于SBA-15載體是不可還原氧化物,故該催化劑上氧氣消耗歸因于活性金屬Ni的氧化消耗。對于Ni/Zr-SBA-15催化劑,其Ni負載量均為5%,可以看到隨著ZrO2的增加,催化劑的氧氣消耗量增加,均大于206.7μmol/g(Ni/SBA-15),表明Ni/Zr-SBA-15催化劑上除活性金屬Ni對氧氣的消耗外,還伴隨著氧空位對氧氣的吸附消耗,這與上述H2-TPR的分析結果相吻合。
圖6 (a)為Ni/Zr-SBA-15催化劑Zr 3d軌道的能譜圖,通過雙峰擬合將該譜圖擬合為四個峰,峰位置分別為181.7、182.5、184.1、184.8 eV,其中,181.7、184.1 eV處的峰歸屬于Zr3+,182.5、184.8 eV處的峰歸屬于Zr4+,譜圖中Zr3+的存在表明ZrO2被部分還原,有氧空位生成[22?24]。對于Ni/Zr-SBA-15催化劑,其O 1s軌道能譜圖與Ni/SBA-5催化劑O 1s軌道能譜圖均只有一個特征峰,為SiO2載體Si?O?Si鍵的特征峰,表明SBA-15載體中Si?O?Si鍵的信號太強,掩蓋了ZrO2中氧原子的能譜信息。因此,為了更加鮮明地區分氧空位與晶格氧特征峰,選用代表性強的Ni/SBA-15催化劑及Ni/ZrO2催化劑進行O 1s軌道特征峰分析。圖6(b)給出了Ni/SBA-15、Ni/ZrO2催化劑O 1s軌道的能譜圖,可以看到兩者O 1s軌道的能譜圖存在明顯差別,對于Ni/SBA-15催化劑,其O 1s軌道的能譜圖有一個明顯的特征峰,電子結合能為532.8 eV,該峰歸屬于SBA-15載體中氧的特征峰[25,26]。對于Ni/ZrO2催化劑,其O 1s軌道的能譜圖有兩個特征峰,電子結合能分別為529.7、531.4 eV,分別歸屬于ZrO2中 的晶格 氧與氧 空位[23,24, 27,28],這 表 明ZrO2的添加可以在催化劑中引入氧空位。
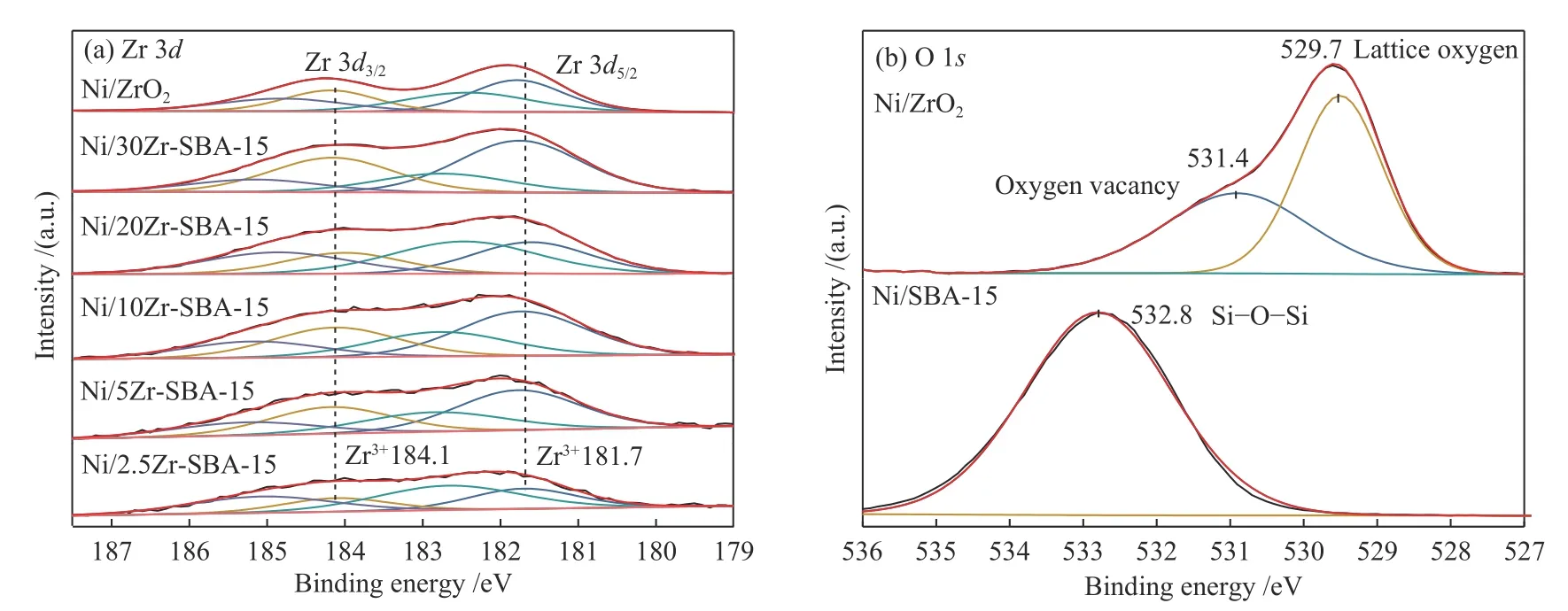
圖6 Ni/Zr-SBA-15催化劑Zr 3d軌道(a)及Ni/SBA-15、Ni/ZrO2催化劑O 1s軌道能譜圖(b)Figure 6 XPS profiles of Ni/Zr-SBA-15 catalysts:Zr 3d (a)and Ni/SBA-15, Ni/ZrO2 catalysts:O 1s(b)
2.2 反應性能評價
圖7 為未改性Ni/SBA-15催化劑上DBF轉化率及產物收率隨反應時間的變化,反應過程中碳平衡均在95%?105%,可以看到反應100 min時DBF完全轉化,反應過程中同時伴隨中間產物的生成及轉化。
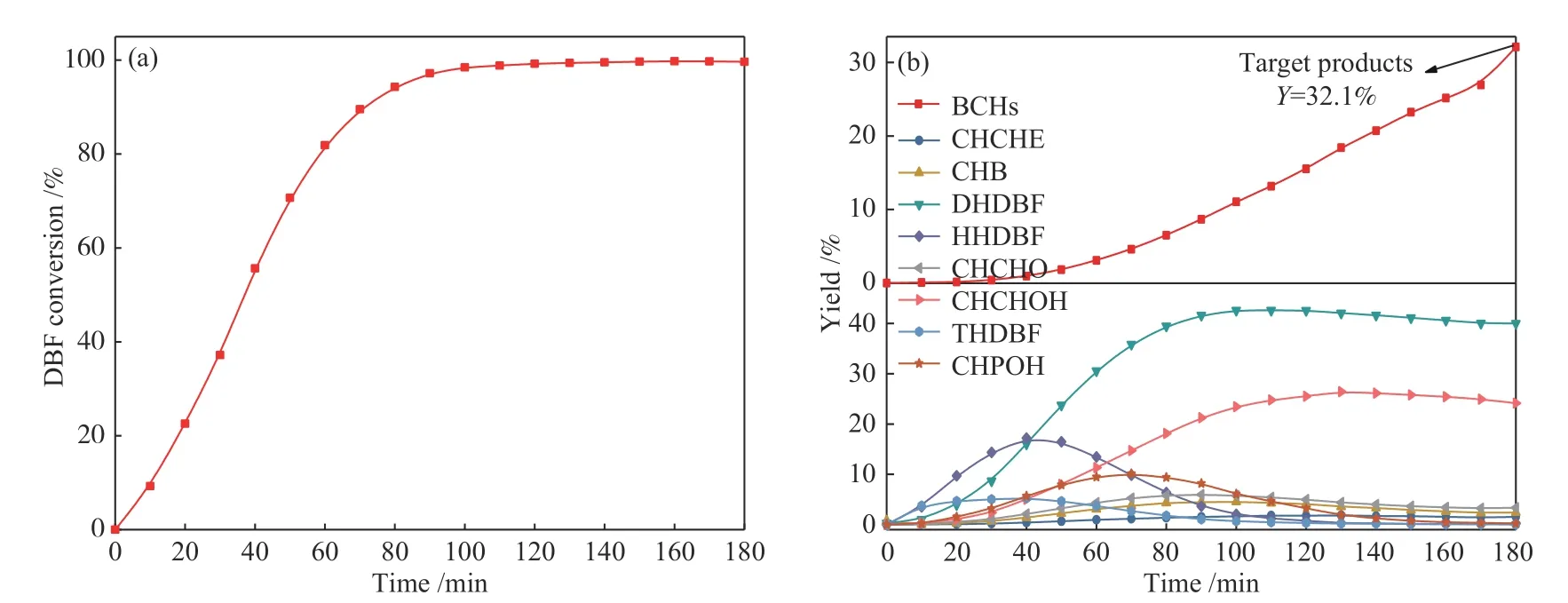
圖7 二苯并呋喃在Ni/SBA-15催化劑上轉化率(a)及產物收率(b)隨時間的變化Figure 7 DBF conversion (a)and product yields(b)over Ni/SBA-15 catalysts as a function of time
從圖7(b)中可以看到,DBF轉化生成四氫二苯并呋喃(THDBF)、六氫二苯并呋喃(HHDBF)、十二氫二苯并呋喃(DHDBF)、2-環己基苯酚(CHPOH)、2-環己基環己醇(CHCHOH)、環己基環己酮(CHCHO)、環己基苯(CHB)、環己基環己烯(CHCHE)、BCHs等物質,其中,THDBF、HHDBF、CHPOH、CHCHO、CHB、CHCHE等物質在反應結束時均完全轉化,而DHDBF、CHCHOH的轉化相對較慢,使得反應結束時DHDBF、CHCHOH、BCHs成為主要的三種反應產物。
根據本實驗及前期DFT計算結果[29,30],作者總結了DBF在Ni催化劑上加氫脫氧的反應網絡,如圖8所示。DBF先加氫生成THDBF、HHDBF,生成的THDBF、HHDBF同時會發生開環反應和加氫反應,高壓下傾向于發生加氫反應生成DHDBF,之后開環生成CHCHOH,同時由THDBF、HHDBF開環生成的CHPOH在高壓下也傾向于發生加氫反應生成CHCHOH,之后CHCHOH再脫羥基生成目標產物BCHs;同時HHDBF也可以通過開環反應再加氫形成環己基環己酮CHCHO,之后CHCHO加氫脫羥基生成BCHs。在DBF加氫脫氧反應網絡中DHDBF發生開環反應的活化能最高,同時CHCHOH在活性金屬Ni表面的吸附能較小,脫氧活化能較高,反應速率較慢,因此,在反應結束時DHDBF、CHCHOH、BCHs有較高的產物收率。
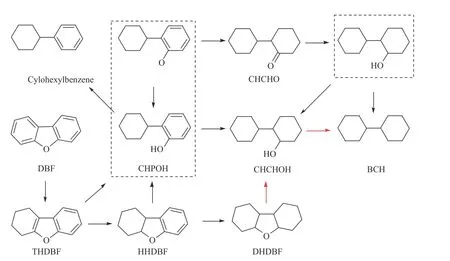
圖8 二苯并呋喃在Ni基催化劑上加氫脫氧反應路徑圖Figure 8 Reaction pathways for HDO of DBF over Ni-based catalysts
在上述Ni/SBA-15催化劑性能評價中,可以發現未改性Ni/SBA-15催化劑斷裂C–O鍵能力不足,反應結束時含氧中間產物DHDBF和CHCHOH均有較高的收率。通過對催化劑進行ZrO2改性,降低含氧中間產物C–O鍵斷裂能壘,以促進反應的進行,為此對改性的Ni/Zr-SBA-15催化劑進行反應評價,并以主要產物DHDBF、CHCHOH、BCHs收率來衡量催化劑性能。
圖9 (a)為Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化劑上DBF轉化率隨反應時間的變化,反應過程中碳平衡均在95%?105%。可以看到,反應結束時三種催化劑上DBF轉化率均為100%,而在反應初始時DBF轉化率存在明顯差異,反應10 min時低轉化率條件下DBF反應速率從大到小依次為Ni/10Zr-SBA-15> Ni/ZrO2> Ni/SBA-15,表 明ZrO2的添加加速了反應物DBF的轉化,促進了反應的進行。
圖9 (b)為DBF加氫脫氧反應過程中Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化劑上主要產物收率隨反應時間的變化。Ni/SBA-15催化劑上目標產物BCHs的收率僅為32%,反應結束時DHDBF、CHCHOH的收率分別高達40%、24%,說明無ZrO2改性的Ni/SBA-5催化劑上金屬Ni本身對含氧中間產物的轉化較慢。在添加10%的ZrO2后,可以看到Ni/10Zr-SBA-15催化劑上主要產物為目標產物BCHs,而含氧中間產物DHDBF、CHCHOH在反應結束后已完全轉化。在反應20 min時兩者的收率達最大值,分別為19%、5.4%,之后快速轉化,這表明ZrO2的添加明顯加快了含氧中間產物的進一步加氫脫氧,提高了目標產物BCHs的收率。對于Ni/ZrO2催化劑,可以看到反應結束時含氧中間產物DHDBF的收率較低,僅為2.7%,含氧中間產物CHCHOH的收率較高,為41%,目標產物BCHs收率為48%,催化活性明顯低于Ni/10Zr-SBA-15催化劑。

圖9 Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化劑上DBF轉化率(a)及主要產物收率(b)隨時間的變化Figure 9 DBF conversion rate (a)and product yields (b)over Ni/SBA-15, Ni/10Zr-SBA-15, Ni/ZrO2 catalysts as a function of time
圖10 為DBF反應速率及目標產物BCHs生成速率隨ZrO2含量的變化。可以看到,隨著ZrO2的增加,兩者均表現出先增加后減小的趨勢。對于未ZrO2改性的Ni/SBA-15催化劑,其DBF反應速率較小,僅為1.25 mmol/(min·g);隨著ZrO2含量的增加,DBF反應速率增加,當ZrO2含量為10%時,DBF反應速率達到最大值,為9.21 mmol/(min·g);當ZrO2添加量大于10%時,DBF反應速率開始下降。同時對比不同催化劑上目標產物BCHs的生成速率,可以發現當ZrO2添加量為20%時BCHs生成速率最大,為3.74 mmol/(min·g),這說明Ni位點與ZrO2之間的協同作用存在最佳匹配,即Ni位點上主要發生H2的吸附解離、DBF及加氫中間產物的吸附和加氫反應,而ZrO2除了促進Ni顆粒的分散外,其提供的氧空位可以促進含氧中間產物上C–O鍵的斷裂。因此,DBF轉化速率主要由暴露的Ni位點決定,而BCHs的生成速率是Ni位點和ZrO2之間的協同匹配決定,從而使得Ni/20Zr-SBA-15催化劑相較Ni/10Zr-SBA-15催化劑DBF反應速率更低,但BCHs生成速率卻更高。DBF反應速率及目標產物BCHs生成速率先增加是因為ZrO2的添加減小了活性金屬Ni的晶粒尺寸,促進了Ni顆粒的分散,同時提供了氧空位,從而加速了反應的進行;當ZrO2含量為30%時DBF反應速率減小,是因為過量ZrO2的添加降低了催化劑的比表面積,同時由于金屬和載體間強相互作用,使得活性金屬Ni周圍部分還原的ZrOx會遷移并覆蓋Ni活性位點[31,32],從而不利于DBF的轉化。

圖10 Ni/Zr-SBA-15催化劑上DBF反應速率、BCHs生成速率隨ZrO2含量的變化Figure 10 DBF reaction rate and BCHs formation rate over Ni/Zr-SBA-15 catalysts
加氫脫氧反應中催化劑酸性對反應活性也有很大的影響,催化劑酸性可以協同活性相促進含氧中間產物發生開環反應以及促進醇脫水反應的發生。在Ni/Zr-SBA-15催化劑酸性分析中,可以發現Ni/Zr-SBA-15催化劑上僅有Lewis酸,且Lewis酸量隨ZrO2的增加表現出先增大后減小的趨勢,Ni/10Zr-SBA-15催化劑上Lewis酸酸量最大。催化劑上的Lewis酸包括配位不飽和的金屬陽離子和氧空位,氧空位能夠吸附含氧化合物,促進含氧化合物中C–O的活化、斷裂[14,33]。因此,分析了Ni的加氫性能(金屬分散度)和氧空位對于產物選擇性的關聯,以進一步確認Ni金屬分散度和氧空位在反應過程中的作用機理。
圖11 為主要產物DHDBF、CHCHOH、BCHs及部分中間產物選擇性與ZrO2含量、Ni分散度及氧空位濃度變化的關系圖。可以看到,對于THDBF、HHDBF、CHPOH,其選擇性在Ni/SBA-15催化劑上最高,之后隨著ZrO2的添加催化劑上Ni分散度提高、氧空位濃度增加,這幾種產物的選擇性隨Ni分散度、氧空位濃度的升高而逐漸降低,表明THDBF、HHDBF、CHPOH的轉化被促進。對于DHDBF、CHCHOH,可以看到其選擇性隨ZrO2含量、Ni分散度、氧空位濃度表現出先增加后減小的趨勢,選擇性在Ni/2.5Zr-SBA-15催化劑上最高,這是因為當ZrO2少量添加時,由于Ni分散度的提高,更多的Ni活性位點可以將DBF、THDBF、HHDBF(初級中間產物)快速轉化為DHDBF、CHCHOH等次級中間產物;但如前所述,DHDBF、CHCHOH的進一步加氫脫氧反應困難、能壘較高,故少量ZrO2的添加提供的氧空位數量有限,不足以將其迅速進一步轉化為BCHs,故DHDBF、CHCHOH發生累積而體現為Ni/SBA-15上兩者的選擇性更高。隨著ZrO2的進一步添加,催化劑斷裂C–O鍵能力增強,DHDBF、CHCHOH的轉化速率加快,選擇性降低。但是,當ZrO2含量為30%時,過量ZrO2的Ni活性位點的覆蓋效應明顯,導致中間產物選擇性又有上升,同時BCHs選擇性下降。以上結果進一步證實了高分散的Ni顆粒可以與氧空位發生協同作用促進含氧中間產物的轉化,提高目標產物的收率。
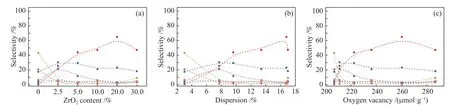
圖 11 Ni/Zr-SBA-15 催化劑上產物選擇性與 ZrO2 含量、Ni 分散度、氧空位濃度的變化Figure 11 The selectivity of products onNi/Zr-SBA-15catalysts varies with Zr O2 content, Ni dispersion, and oxygen vacancyconcentration
3 結 論
ZrO2的添加會提高Ni顆粒分散度、引入氧空位,進而影響加氫脫氧反應的性能。隨著ZrO2的添加,Ni/SBA-15催化劑活性表現出先上升后下降的趨勢,當ZrO2添加量為10%時,二苯并呋喃反應速率最高,為9.21 mmol/(min·g);當ZrO2添加量為20%時聯環己烷生成速率最高,為3.74 mmol/(min·g)。這是因為適量ZrO2的添加增強了Ni與載體間相互作用,促進了Ni顆粒的分散,有利于DBF的轉化,同時ZrO2的添加也在催化劑中引入了氧空位,促進了含氧化合物中C?O鍵的斷裂,有利于加氫脫氧反應的進行;而過量ZrO2的添加降低了催化劑的比表面積,不利于Ni顆粒的分散,不利于反應的進行。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
