熱可逆離子液體-低共熔溶劑雙水相體系的相行為及理化特性研究
2021-06-03 07:39:36張莉莉李艷高靜
化工學報 2021年5期
關鍵詞:體系
張莉莉,李艷,高靜
(1廣東藥科大學食品科學學院,廣東中山528458;2廣東海洋大學食品科技學院,廣東湛江524088)
引 言
雙水相體系(aqueous biphasic systems)分離萃取技術歷史悠久,具有條件溫和、生物相容性高、無有機溶劑殘留、可連續操作和易于放大等優點,在食品、醫藥等領域應用廣泛。然而,傳統聚合物雙水相體系黏度大、選擇性低,且不利于萃取分子的分離純化。離子液體(ionic liquids,ILs)具有可設計性,以咪唑類為代表的離子液體溶解能力強、熱化學性質穩定、可循環利用,是理想的溶劑和反應介質[1-2]。2003年,Gutowski等[3]利用咪唑基離子液體與K3PO4構建雙水相體系。后來,以不同種類的離子液體為chaotropic組分、以無機鹽或有機鹽為kosmotropic組分構建的雙水相體系不斷發展。低共熔溶劑(deep eutectic solvents,DESs)是由氫鍵供體(HBD)和氫鍵受體(HBA)[4]或電子給體與電子受體[5-7]通過非共價相互作用所形成的共晶混合物,具有與離子液體相近的物化性質,可以設計成低毒或綠色的溶劑體系,而且具有廉價易得和可生物降解的優勢。Zeng等[8]用氯化膽堿和尿素形成的低共熔溶劑與K2HPO4構建新型雙水相體系并成功提取牛血清白蛋白。近年來,離子液體雙水相體系和低共熔溶劑雙水相體系在蛋白質[9-10]、核酸[11-12]等生物分子的萃取領域中發展迅速。但是,以K3PO4、K2HPO4為代表的kosmotropic鹽形成的雙水相體系多表現出強堿性。不但腐蝕萃取設備,而且不利于保持生物分子穩定性和生物活性,引起研究者們的重視。
溶劑體系的化學平衡與溫度、組分的性質和溶質的性質密切相關,理解萃取過程或化學反應最有效的途徑是考察介質的物化性質[13]。因此,設計一種適用于生物分子萃取的雙水相體系需要考慮體系結構、溶液pH以及環境溫度等因素[14-16]。其中,溫度對雙水相體系的作用較為復雜。早期,Zafarani-Moattar等[17]指出溫度對雙水相體系沒有顯著的影響。因此,在萃取生物分子的許多研究中也忽略了對溫度的控制[18-19]。然而,Sadeghi等[20]提出離子液體-鹽雙水相的成相能力隨溫度的升高而減弱,表現為具有高臨界共熔溫度(upper critical solution temperature,UCST)的相轉變行為。這一結論得到了眾多學者的大力支持[21]。UCST型離子液體雙水相體系已在分離蛋白質[22-23]、酶[24]等方面獲得廣泛肯定。近年來,研究發現,三丁基辛基溴化([P4448]Br)與K3PO4構成的雙水相成相能力隨溫度升高而增強,表現為具有低臨界共熔溫度(lower critical solution temperature,LCST)的相轉變行為[25]。UCST和LCST型離子液體-鹽雙水相體系的建立成為重新審視溫度對離子液體雙水相體系影響作用的重要突破口。
本文以氯化膽堿(ChCl)和碳水化合物分別作HBA和HBD合成環境友好的低共熔溶劑,以此替代無機鹽/有機鹽與離子液體形成一種溶液環境溫和的雙水相體系。通過考察不同離子液體-低共熔溶劑雙水相體系相行為隨溫度變化的規律,篩選出具有UCST型和LCST型相轉變行為的兩類雙水相體系,系統研究了熱可逆離子液體-低共熔溶劑-水三元體系的黏度、密度、電導率以及pH與溫度的相關性,并利用量子化學計算從微觀角度分析離子液體、低共熔溶劑與分子之間的相互作用,旨在揭示離子液體-低共熔溶劑雙水相體系形成的機理,為雙水相體系在綠色萃取領域的應用提供理論依據。
1 實驗材料和方法
1.1 材料
1-丁基-3-甲基咪唑四氟硼酸鹽([C4mim]BF4)、1-丁基-3-甲基咪唑溴鹽([C4mim]Br)、1-丁基-3-甲基咪唑氯鹽([C4mim]Cl)、1-丁基-3-甲基咪唑三氟乙酸鹽([C4mim]CF3COO)、四丁基四氟硼酸([P4444]BF4)、四丁基溴化([P4444]Br)、四丁基氯化([P4444]Cl)、四丁基三氟乙酸([P4444]CF3COO)、三丁基辛基四氟硼酸([P4448]BF4)、三丁基辛基溴化([P4448]Br)、三丁基辛基氯化([P4448]Cl)、三丁基辛基三氟乙酸([P4444]CF3COO)、四丁基四氟硼酸銨([N4444]BF4)、四丁基溴化銨([N4444]Br)、四丁基氯化銨([N4444]Cl)、四丁基三氟乙酸銨([N4444]CF3COO)(純度均≥98%),中國科學院蘭州化學物理研究所;氯化膽堿(ChCl)(純度>98%)、D-果糖(Fru)(純度>99%),上海Aladdin化學試劑公司;D-核糖(Rib)(純度>99%),源葉生物有限公司;D-葡萄糖(Glu)(分析純)、蔗糖(Sur)(分析純),廣東光華有限公司;4-硝基苯胺(NA),北京百靈威科技有限公司;N,N-乙基-4-硝基苯胺(DENA),Fluorochem有限公司;甜菜堿30(RD),Sigma-Aldrich有限公司。離子液體及低共熔溶劑化學結構如圖1所示。
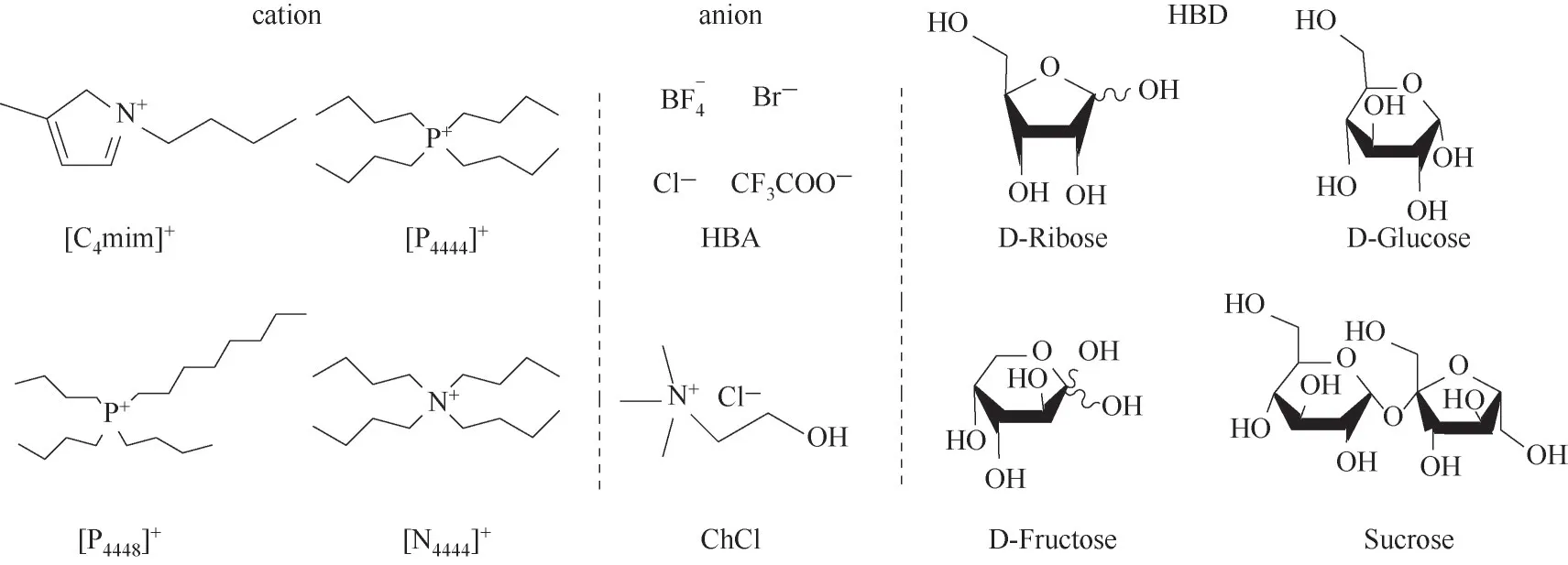
圖1 離子液體及低共熔溶劑的化學結構Fig.1 Chemical structure of ionic liquid sand deep eutectic solvents
1.2 儀器與設備
HAAKEMARSIII模塊化高級流變儀,德國賽默飛世爾科技;Cary60紫外可見分光光度計,安捷倫有限公司;STA449F3同步熱分析儀,德國耐馳儀器制造有限公司;DDSJ-308A電導率儀,上海儀電科學儀器股份有限公司;PHSJ-3FpH計,上海儀電科學儀器股份有限公司;KQ-300超聲波清洗器,上海精科實業有限公司;HH-6數顯電子恒溫水浴鍋,常州國華電器有限公司;AW120電子天平,日本島津公司。
1.3 實驗方法
1.3.1 離子液體和低共熔溶劑基本性質測定 水分測定采用GB5009.3—2016測定方法,電熱恒溫鼓風干燥箱101.3 kPa,378.15 K。黏度測定:模塊化高級流變儀,298.15 K、直徑20 mm、間隙1 mm平行幾何板結構,平衡樣品溫度2 min、1 s-1剪切速率測定30 s。極性測定[26-28]:紫外可見分光光度計298.15 K,NA、DENA、RD、甲醇、離子液體的Kamlet-Taft極性參數和極性指數由式(1)~式(6)計算。熱重分析[29]:同步熱分析儀N2,298.15~973.15 K,進樣量4~6 mg,加熱速率10 K·min-1,吹掃氣流20 cm3·min-1。所有的樣品進行三次重復測定。

式中,vmax(dye)是染料的最大波數,cm-1;λmax(dye)是染料的最大吸收波長,nm;ET(30)是甜菜堿30染料的極性指數,kcal·mol-1(1 kcal=4185.85 J);ENT是以水(ENT=1.00)和四甲基硅烷(ENT=0.00)為參比溶劑進行無量綱標準化的值,kcal·mol-1;ET(solvent)為溶劑的極性指數,kcal·mol-1;α是氫鍵酸性;β是氫鍵堿性;π*是偶極子/極化率。
1.3.2 相圖繪制 雙水相體系相圖采用“濁點滴定”法繪制[6]。準確稱取1 g離子液體水溶液(6∶4,質量比)于試管,置于恒溫水浴鍋(298.15、308.15、318.15、328.15 K),隨后向試管中逐滴加入低共熔溶劑水溶液(8∶2,質量比),至混合體系由澄清變為混濁,記錄各組分數值。再逐滴加入水至體系變澄清,記錄各組分數值。如此反復,直至形成與單相狀態相對應的清澈透明的溶液[6]。計算并記錄各組分的質量百分數,繪制雙水相體系相圖。利用式(7)中的數學方法擬合離子液體-低共熔溶劑雙水相體系數值從而描述和關聯離散點組與坐標之間的函數關系[10]。
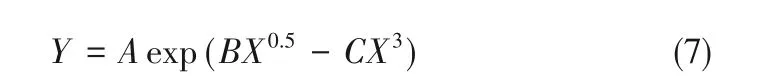
式中,Y是離子液體的質量分數,%;X是低共熔溶劑的質量分數,%;A、B、C是用最小二乘法回歸得到的擬合參數。
1.3.3 三元混合體系的理化特性測定 以質量比
為0.30∶0.10∶0.60配制8種溫敏性離子液體-低共熔溶劑-水溶液,測定了三元體系在298.15~338.15 K下的黏度、密度、電導率和pH變化規律,記錄測定數值[30]。所有的樣品進行三次重復測定。
1.3.4 量子化學計算 分別構建[P4448]Cl、ChCl、Fru、H2O的三維結構,采用Molclus分別搜索初始構象,體 系 有:[P4448]Cl、[P4448]Cl-H2O、ChCl、[ChCl][Fru]、
[ChCl][Fru]-H2O(1∶1)、[P4448]Cl-[ChCl][Fru]-H2O(1∶1∶1)[31]。使用MOPAC程序中的PM7半經驗方法優化初始結構,采用能量(0.5 kcal·mol-1)和構象差異[0.25 ?(1?=0.1 nm)]方法進行聚類[32]。采用DFT方法在B3LYP-D3/6-31G*水平下進行結構優化,對最后優化的前3個構象進行單點能計算,計算水平為B3LYP-D3/def2-TZVP,按照能量進行排序,取能量最低的構象計算相互作用能[33-34]。計算時添加基組重疊誤差(basis set superposition error,BSSE)較正[35-36]。最終結合能采用式(8)計算:
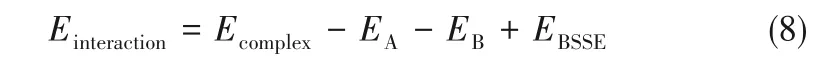
式中,Einteraction是復合物的能量,kJ·mol-1;EA和EB分別為兩個相互作用組分的能量,kJ·mol-1;EBSSE表示BSSE較正能,kJ·mol-1。
2 實驗結果與討論
2.1 離子液體、低共熔溶劑的基本性質比較
根據“液體孔洞”理論,水分子與kosmotropic低共熔溶劑形成配合物,引起水分子氫鍵網絡空腔表面張力增加,導致chaotropic離子液體與水分子作用減弱,從而形成相分離。但是,由不同陰、陽離子排列組合形成的離子液體數目達1018,不同HBA和HBD組合也可以形成數量龐大的低共熔溶劑系列。Zafarani-Moattar等[37]通過測定由氯化膽堿和蔗糖合成的低共熔溶劑與聚乙二醇400構建的體系的密度和聲速,指出該體系與半理想溶液存在負偏差,不利于溶劑-溶劑相互作用,從而導致相分離。因此,利用離子液體和低共熔溶劑的雙重“可設計性”,探索雙水相體系組分的基本性質是構建和設計離子液體-低共熔溶劑雙水相體系的核心。
表1列舉了16種離子液體和4種低共熔溶劑的黏度。整體來看,離子液體和低共熔溶劑的黏度均明顯大于乙醇等有機試劑的黏度,而且低共熔溶劑的黏度普遍顯著高于離子液體。其中,黏度最大的離子液體是[P4448]Cl,黏度最小的低共熔溶劑是[ChCl][Rib]。在298.15 K下[ChCl][Rib]的黏度為(23.40×103±0.25×103)mPa·s,是[P4448]Cl的18.57倍,而乙醇的黏度只有1.074 mPa·s。黏度大導致傳質困難,進而抑制了生物分子的提取效率,這是離子液體和低共熔溶劑在萃取應用中面臨的共同難題。
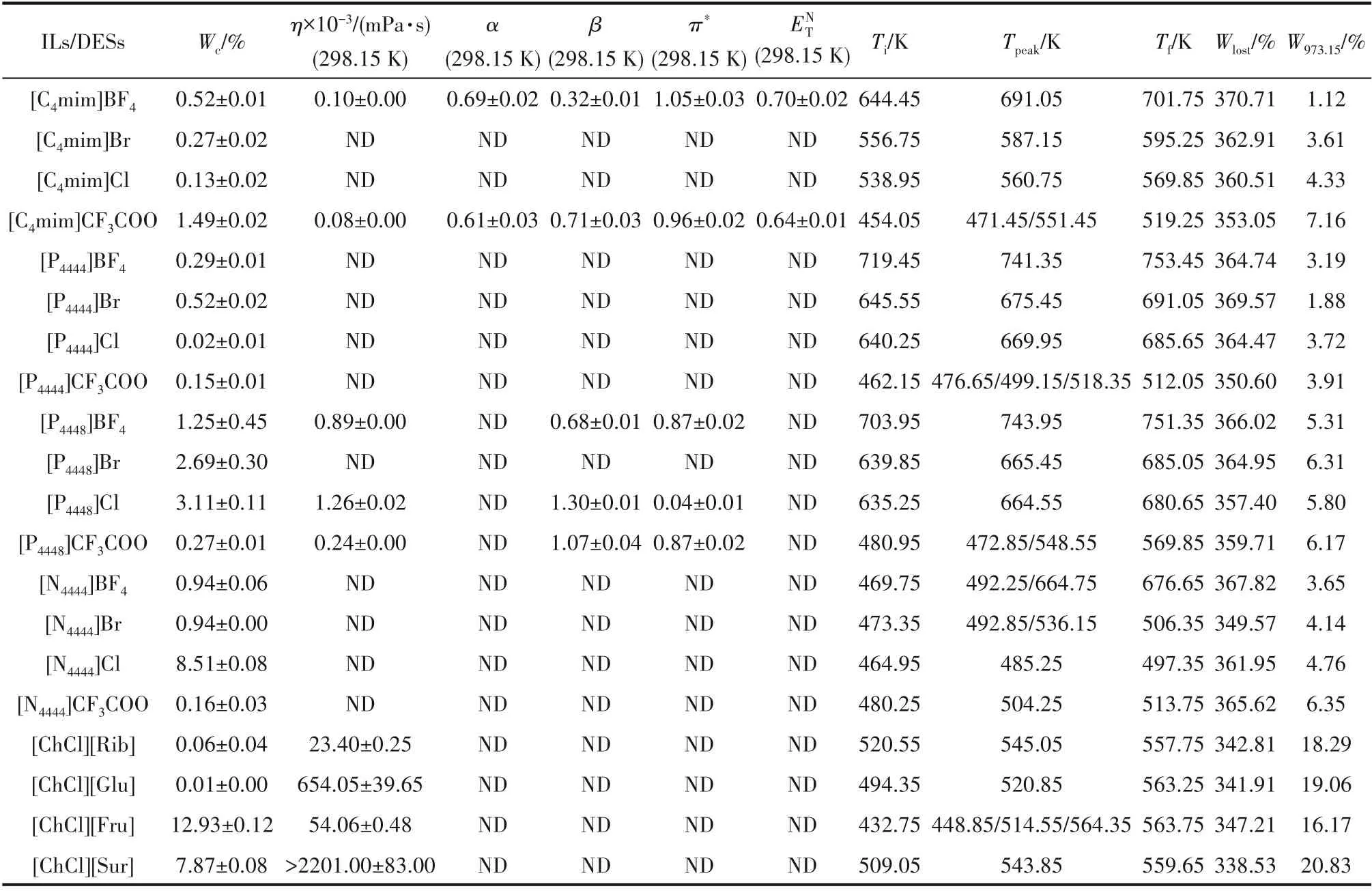
表1 離子液體及低共熔溶劑的基本性質Table 1 Properties of ionic liquids and deep eutectic solvents
不過,[C4mim]CF3COO和[ChCl][Fru]的最低降解起始溫度分別為454.05和432.75 K,兩者所表現出的良好熱穩定性為構建萃取體系提供了較大的溫度范圍選擇空間。在利用程序控制溫度的過程中,離子液體和低共熔溶劑在加熱過程中發生升華、氣化、分解出氣體或失去結晶水等物理現象,質量逐漸減小。由表1可知,整體上看,離子液體較低共熔溶劑表現出更高的降解起始溫度(Ti)和降解終止溫度(Tf),但是,與離子液體相比,低共熔溶劑的主要質量損失(Wlost)較低,973.15 K下的殘余質量(W973.15)更大,說明低共熔溶劑的熱穩定性更好。例如,W973.15最大的離子液體是[C4mim]CF3COO,最小的低共熔溶劑是[ChCl][Fru],[ChCl][Fru]的殘余質量(W973.15=16.17%)是[C4mim]CF3COO(W973.15=7.16%)的2.26倍。
極性是描述離子液體、低共熔溶劑對水分子以及萃取目標分子溶解能力的重要參數。離子液體往往同時具有疏水、不飽和鍵、氫鍵供體、氫鍵受體、靜電等多種結構片段,可與溶質之間發生疏水、氫鍵、靜電、π-π等多種相互作用[38]。低共熔溶劑的HBD和HBA之間不僅存在氫鍵作用,還有C—H…O和C—H…π相互作用[39]。由于純低共熔溶劑黏度過大,無法測定極性。表1列舉了在298.15 K下測定的離子液體的Kamlet-Taft探針行為和極性指數。一般情況下,具有較小β值的咪唑類離子液體形成氫鍵的能力更強。例如,與[C4mim]CF3COO(0.71±0.03)相比,[C4mim]BF4(0.32±0.01)酸性顯著,具有更強的氫鍵形成能力。季鹽離子液體的β值規律為:[P4448]Cl>[P4448]CF3COO>[P4448]BF4。由此可見,相同陽離子的離子液體其陰離子氫鍵堿性變化遵循:Cl->CF3COO->BF4-。
2.2 離子液體-低共熔溶劑雙水相體系的相行為
2.2.1 溫度的影響 生物活性分子對溫度十分敏感,因此明確溫度對離子液體雙水相萃取體系穩定性的影響至關重要。但是,溫度對離子液體雙水相體系的作用始終存在爭議,隨著離子液體雙水相體系種類的增加,情況也越來越復雜。Passos等[40]構筑了一種對溫度敏感的熱可逆離子液體-聚合物雙水相體系,而且明確指出低溫利于該體系的形成。許多研究表明低溫有利于離子液體-鹽雙水相體系的形成,表現出具有UCST型的相轉變行為[41]。Griffin等[42]觀察到在聚乙二醇-鹽雙水相中,kosmotropic鹽濃度增加與溫度升高在促進雙水相體系熵貢獻的過程中具有類似的效果,即通過升高溫度增加相的不混溶性從而擴大兩相區域面積,表現為LCST型雙水相體系。Gao等[25]最新研究發現:由三丁基辛基溴化([P4448]Br)構成的離子液體-鹽雙水相的成相能力隨著溫度的升高而增強,也表現出具有LCST型的相轉變行為。
本文測定了16種離子液體與4種低共熔溶劑在298.15~328.15 K的相圖,篩選出3種UCST型和19種LCST型離子液體-低共熔溶劑雙水相,結果見表2。其中,僅有[C4mim]BF4能形成UCST型雙水相體 系 體 系,而[P4444]Br、[P4444]Cl、[P4448]Br、[P4448]Cl、[N4444]CF3COO能形成LCST型雙水相體系。因此,離子液體的陽離子結構與其熱致相變的能力密切相關。圖2列舉了分別以[C4mim]BF4和[N4444]CF3COO為代表的UCST型[圖2(a)]和LCST型[圖2(b)]雙水相。類似地,Schaeffer等[43]指出[P44414]Cl-HCl表現為LCST型雙水相。由此可見,離子液體的種類是決定溶劑體系熱誘導相轉變類型的關鍵因素。
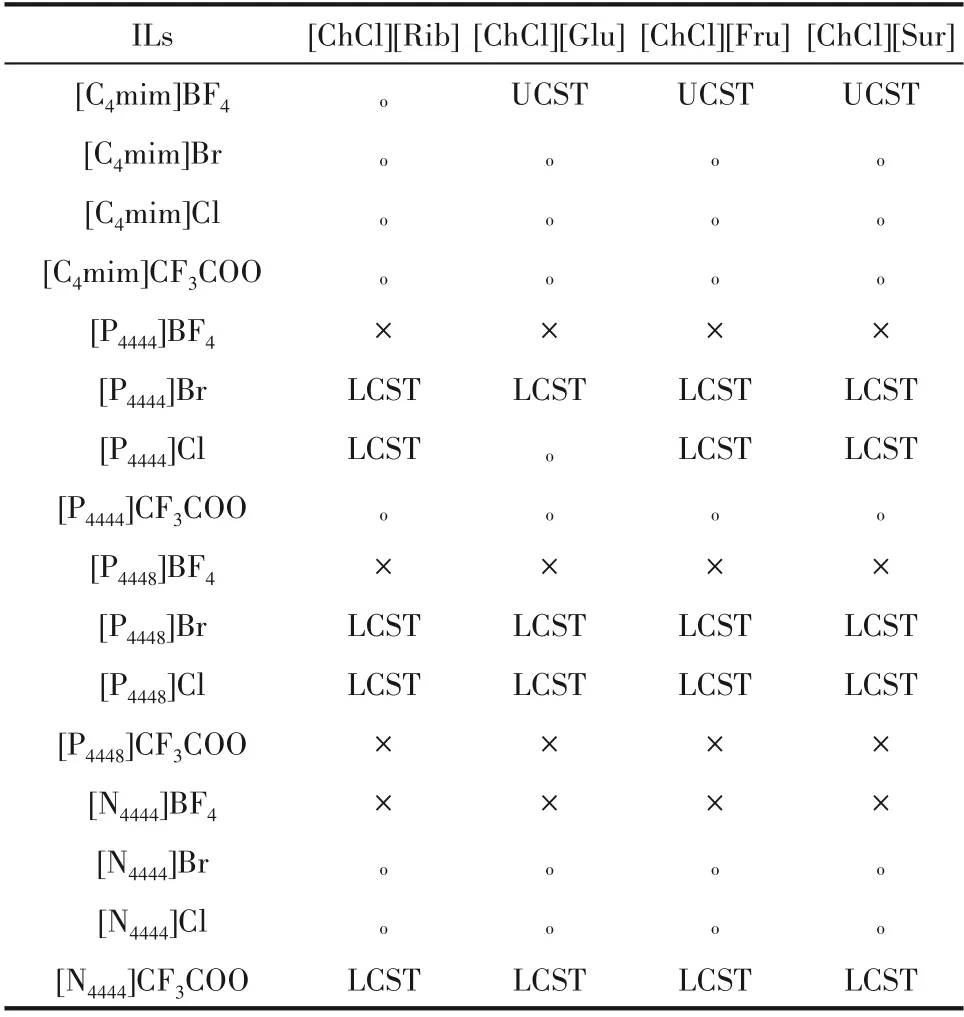
表2 熱可逆離子液體-低共熔溶劑-水體系的構建Table 2 Construction of thermo reversible ILs-DESs-H 2O system
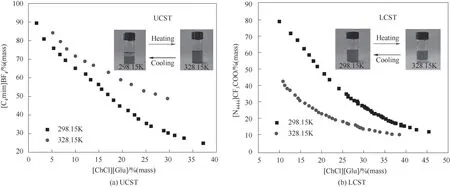
圖2 UCST型(a)與LCST型(b)離子液體-低共熔溶劑體系的雙水相相圖Fig.2 Phase diagrams of UCST(a)and LCST(b)of IL-DES-H2O
2.2.2 離子液體的影響 研究表明β值介于0.38~0.60之間的咪唑類離子液體具有良好的成相能力[44]。例如,較高氫鍵堿性的[C4mim]Cl(β=0.87,數據來自文獻[44])和[C4mim]CF3COO(β=0.71±0.03)因親水性強而不利于雙水相的形成,而[C4mim]BF4具有適中的氫鍵堿性(β=0.32±0.01),更易形成雙水相體系。圖3對比了308.15 K下6種離子液體分別與低共熔溶劑[ChCl][Fru]和[ChCl][Sur]構建的雙水相體系的成相能力。可以看出,離子液體成相能力的規律為[P4448]Br>>[P4448]Cl>[N4444]CF3COO>[C4mim]BF4>[P4444]Br>[P4444]Cl。與陰離子相比,離子液體的陽離子種類對其成相能力的影響更為顯著,其成相能力的差異性主要表現在陽離子電荷分布不同[23]。[P4448]+具有較長的側鏈,與水相互作用的自由能大,分子有逃逸水相的傾向,導致水溶液摩爾體積增大,因此成相能力大[45]。陰離子對成相能力的影響規律為:Br->Cl-。在季基/季銨基離子液體與磷酸鉀形成雙水相體系的研究中也有相似的規律[46]。因為Br-可與陽離子形成較強的庫侖力,從水中接受質子能力比Cl-弱,利于相分離[47]。
2.2.3 低共熔溶劑的影響 糖類物質作為HBA與氯化膽堿合成的低共熔溶劑因穩定性強、廉價易得
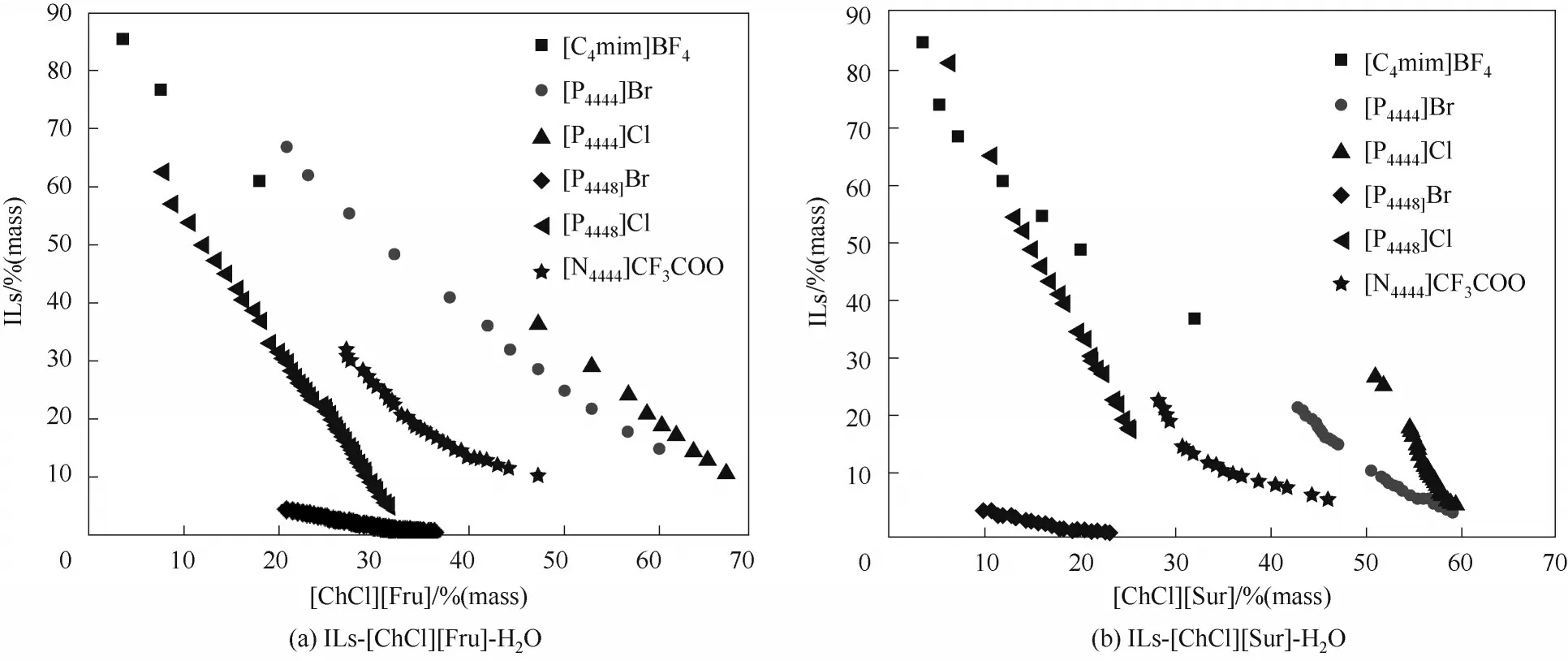
圖3 離子液體-[ChCl][Fru]和離子液體-[ChCl][Sur]在308.15 K下的雙水相相圖Fig.3 Phase diagrams of ILs-[ChCl][Fru]-H2Oand ILs-[ChCl][Sur]-H2Oat 308.15 K
且綠色的特點常用于雙水相體系的構建和生物分子的萃取。如圖4所示,低共熔溶劑與離子液體形成雙水相的能力大小為:[ChCl][Sur]>[ChCl][Glu]>[ChCl][Fru]>[ChCl][Rib]。蔗糖具有較多的氫鍵結合位點,可形成更多的交聯結構,與水分子之間存在較強的氫鍵作用,有利于與離子液體競爭水分子,因此其相形成能力更強。另一方面,結合表1中的黏度數據發現,低共熔溶劑的黏度大小影響雙水相體系兩相間的物質傳遞,是設計雙水相體系的重要參考因素。例如,[ChCl][Sur]的黏度最大[大于(2201.00×103±83.00×103)mPa·s],而[ChCl][Rib]的 黏度最小[(23.40×103±0.25×103)mPa·s],其黏度大小與成相能力規律完全一致。
2.2.4 雙水相體系數學模型 利用經驗公式(7)對
本文所有雙水相體系的雙節線相圖進行擬合,相關參數見表3。圖5列舉了308.15 K下[P4444]Br-[ChCl][Fru]雙水相體系雙節線擬合的結果。結果表明離子液體-低共熔溶劑雙水相體系離散點組數值符合方程擬合模型。通過在統計學范圍內擬合數據得到線性回歸分析,并以最小二乘法進行參數估計,為雙水相體系的工業化應用提供重要的設計依據。
2.3 離子液體-低共熔溶劑-水三元體系的理化性質
2.3.1 黏度 針對表2構建的熱可逆UCST型和LCST型離子液體-低共熔溶劑雙水相體系,測定了離子液體-低共熔溶劑-水(0.30∶0.10∶0.60)三元體系的理化性質的變化情況(圖6)。圖6(a)是三元體系黏度隨溫度的變化趨勢。結果表明,其黏度高度依賴溫度,且呈指數變化,實驗數據通過式(9)擬合處理。溫度升高加快分子振動,引起黏聚性降低,導致黏度減小[48]。純離子液體黏度(表1)較離子液體-低共熔溶劑-水三元體系[圖6(a)]黏度顯著降低。除去固體狀態未測定的離子液體,三元體系與單獨組分黏度大小規律一致。低共熔溶劑對三元體系黏度影響不大,而相同[ChCl][Fru]條件下,三元體系黏 度 變 化 規 律 為[P4448]Cl>[P4444]Cl>[N4444]CF3COO>[P4444]Br>[C4mim]BF4。表明離子液體種類是影響三元體系黏度變化的主要因素。黏度取決于流體的分子結構,高度依賴于組成離子之間的相互作用:靜電,范德華力和氫鍵[49]。水分子破壞了離子液體陰、陽離子的鏈結構,降低相互之間的庫侖作用,使其分離出陰陽離子殼層,最終導致黏度降低[50]。
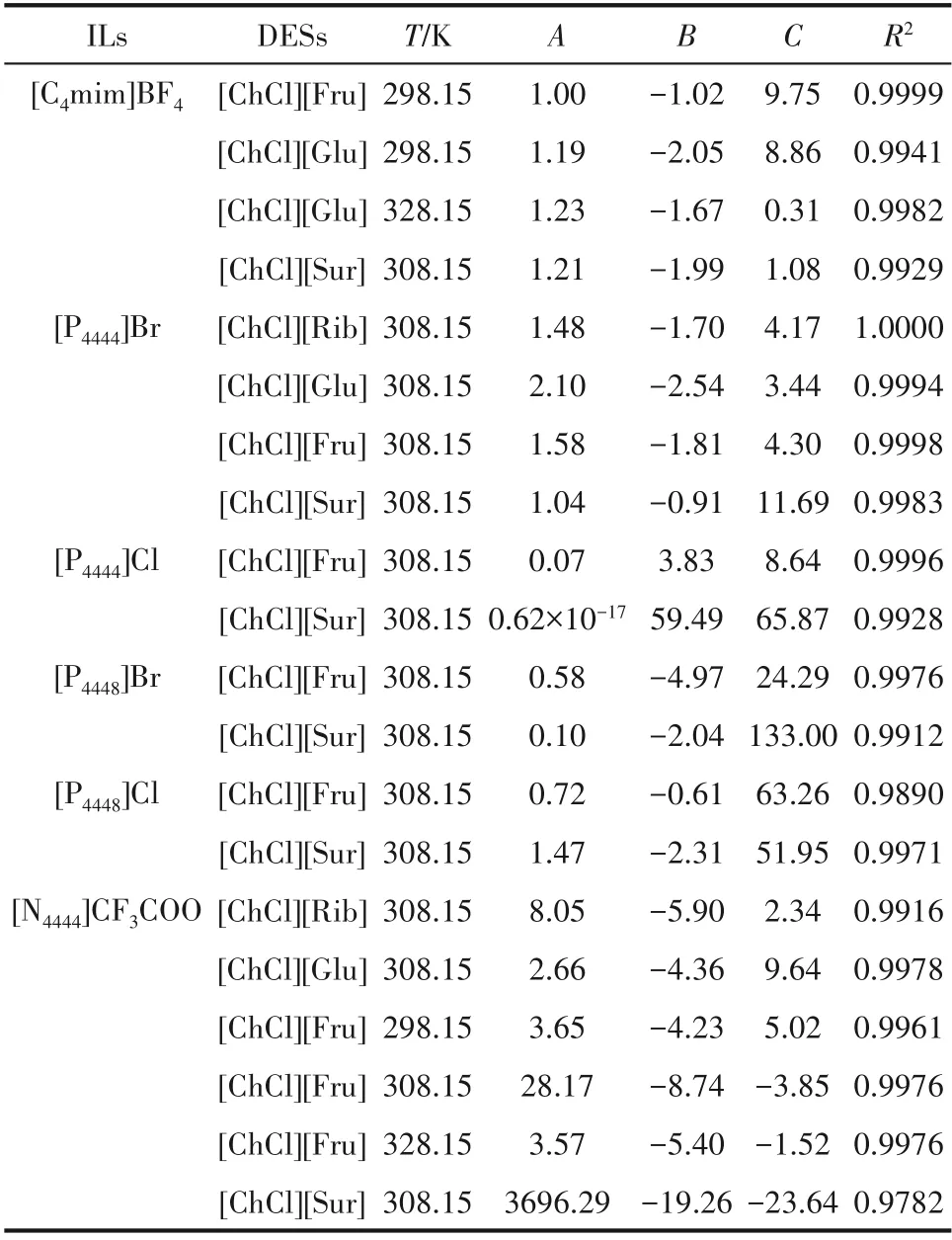
表3 離子液體-低共熔溶劑體系雙水相相圖實驗數據相關性擬合Table 3 Phase diagram of ILs-DESs-H 2O with the corresponding fitting of the experimental data
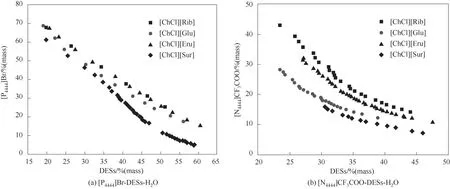
圖4 [P4444]Br-低共熔溶劑(a)和[N4444]CF3COO-低共熔溶劑(b)在308.15 K下的雙水相相圖Fig.4 Phase diagrams of DESs-[P4444]Br-H2O(a)and DESs-[N4444]CF3COO-H2O(b)at 308.15 K
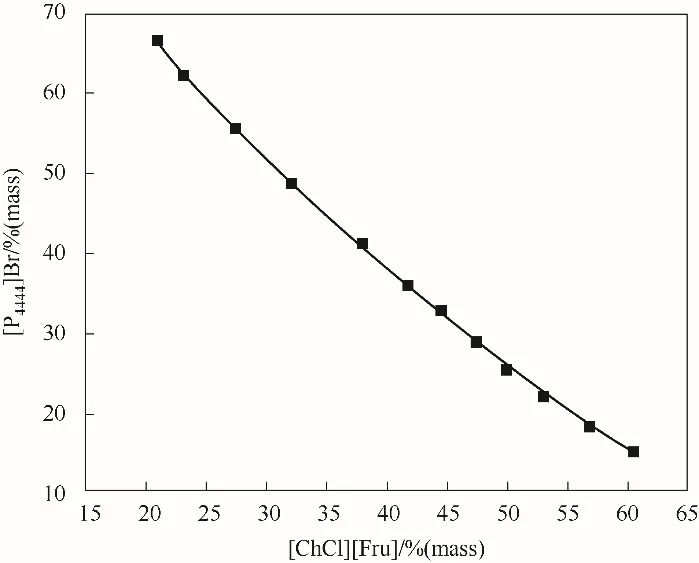
圖5 308.15 K下[P4444]Br-[ChCl][Fru]雙水相相圖實驗數據相關性擬合Fig.5 Phase diagramof[P4444]Br-[ChCl][Fru]-H2Owith the corresponding fitting of the experimental data at 308.15 K
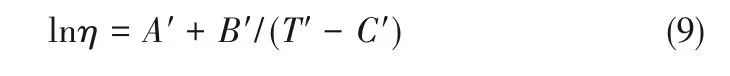
式中,η是三元體系黏度,mPa·s;T為三元體系溫度,K;A′、B′、C′是擬合參數。
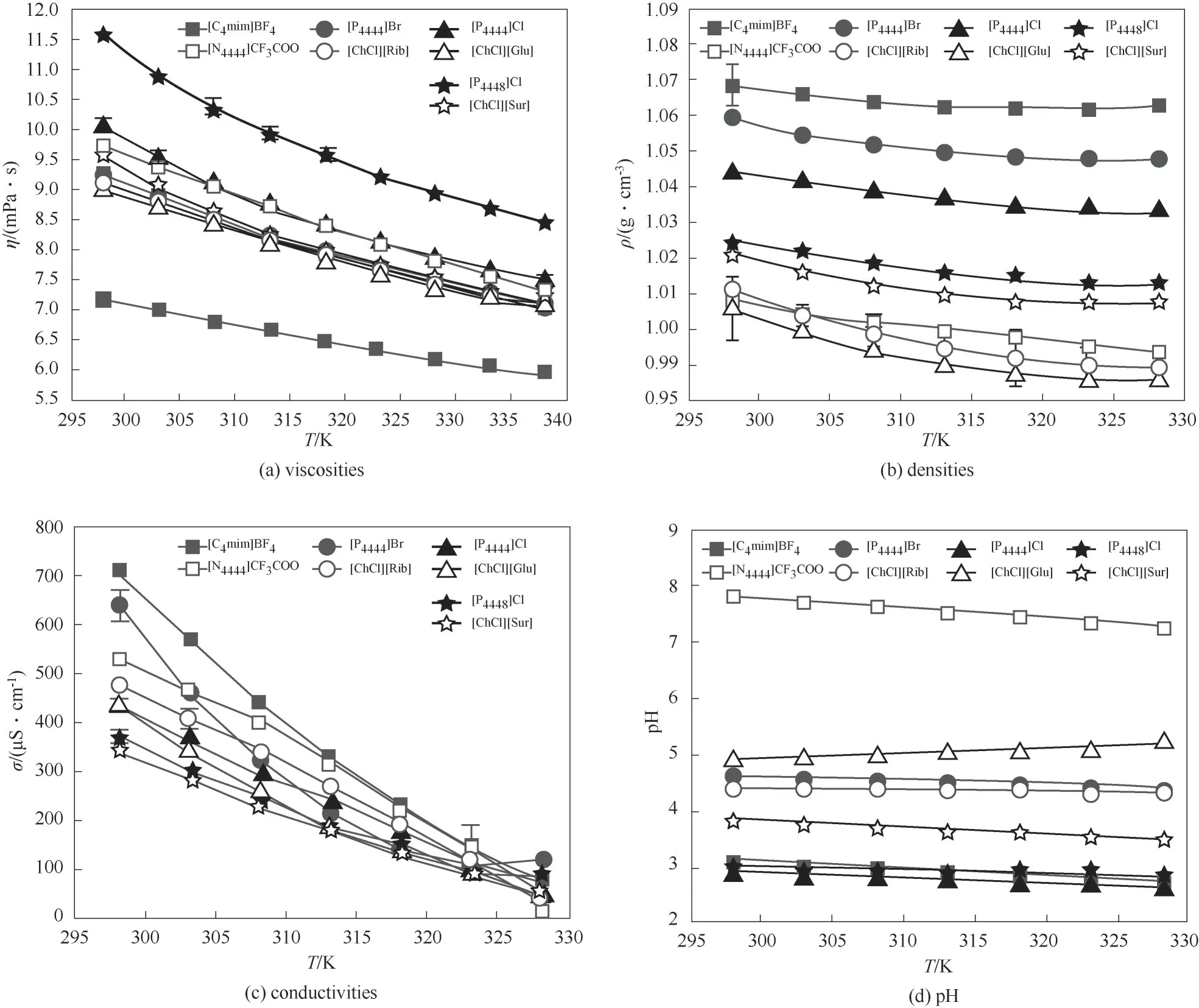
圖6 不同溫度下離子液體-低共熔溶劑-水三元體系黏度、密度、電導率和pH的變化趨勢Fig.6 Viscosities,densities,conductivities,pH for ILs-DESs-H 2Oas a function of temperature
2.3.2 密度 密度是溫度的函數,一般來說,體系密度隨環境溫度升高逐漸降低。圖6(b)測定了不同溫度下離子液體-低共熔溶劑-水三元體系密度變化,實驗數據經式(10)擬合處理。結果表明,由不同離子液體構成的雙水相其密度關系為[C4mim]BF4>[P4444]Br>[P4444]Cl>[P4448]Cl>[N4444]CF3COO。離子液體的陽離子是影響密度的重要因素。季鹽離子液體的陽離子烷基側鏈鏈長增加,密度減小。Bounsiar等[51]研究發現1-丁基-雙酰亞胺吡啶比1-己基-雙酰亞胺吡啶的密度大,也是因為陽離子上的烷基鏈鏈長增加,分子摩爾體積變大,導致密度降低。另外,陰離子的密度與分子量呈正比,含Br-(79.904)的陰離子比含Cl-(35.453)的密度大。
如圖7,通過對密度數據進行式(11)~式(14)的處理,得到標準摩爾體積(Vm)、分子體積(V)、標準熵(S?)等相關參數[52]。可以看出,標準摩爾體積與標準摩爾熵隨溫度升高而增大,而晶格能隨溫度升高逐漸降低。標準摩爾體積與體系內部分子間距密切相關,當溫度升高,體系內分子熱運動加快,分子間締合作用減弱,分子間距增大,體系標準摩爾體積增加[圖7(a)]。另外,溫度升高導致體系內微粒混亂,標準熵增大[圖7(b)]。計算三元體系的晶格能,可以獲得其穩定性的相關信息。與NaCl(786 kJ·mol-1)相比,三元體系內陽離子的不對稱性以及體積大的性質增大了空間位阻,減少了陰陽離子的相互作用,增大了晶格穩定性[圖7(c)]。但是,溫度的增加增強了體系內部離子之間的相互作用,三元體系的晶格穩定性降低。
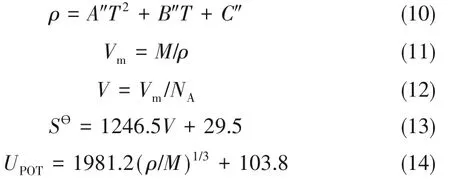
式中,ρ為三元體系密度,g·cm-3;T為三元體系溫度,K;Vm為標準摩爾體積,cm3·mol-1;M為摩爾質量,g·mol-1;V為分子體積,cm3;NA為Avogadro常數,mol-1;S?為標準摩爾熵,J·K-1·mol-1;UPOT為晶格能,kJ·mol-1;A″、B″、C″是擬合參數。
2.3.3 電導率 為明確三元體系受溫度影響內部電荷流動的難易程度,測定了混合溶液電導率值隨溫度變化的基礎數據,實驗結果通過式(15)擬合。如圖6(c),三元體系電導率隨溫度增大逐漸降低,離子液體種類是引起電導率變化的關鍵因素,這是因為由離子鍵結合的離子液體溶于水且在加熱狀態下解離成自由移動的離子,而不同離子液體解離后的電解質導電能力具有差異性。另外,具有相同陰離子[Cl]-的離子液體陽離子烷基鏈長度增加,電導率降低。烷基鏈長度增加了液相中的分散相互作用,導致離子遷移困難[53]。
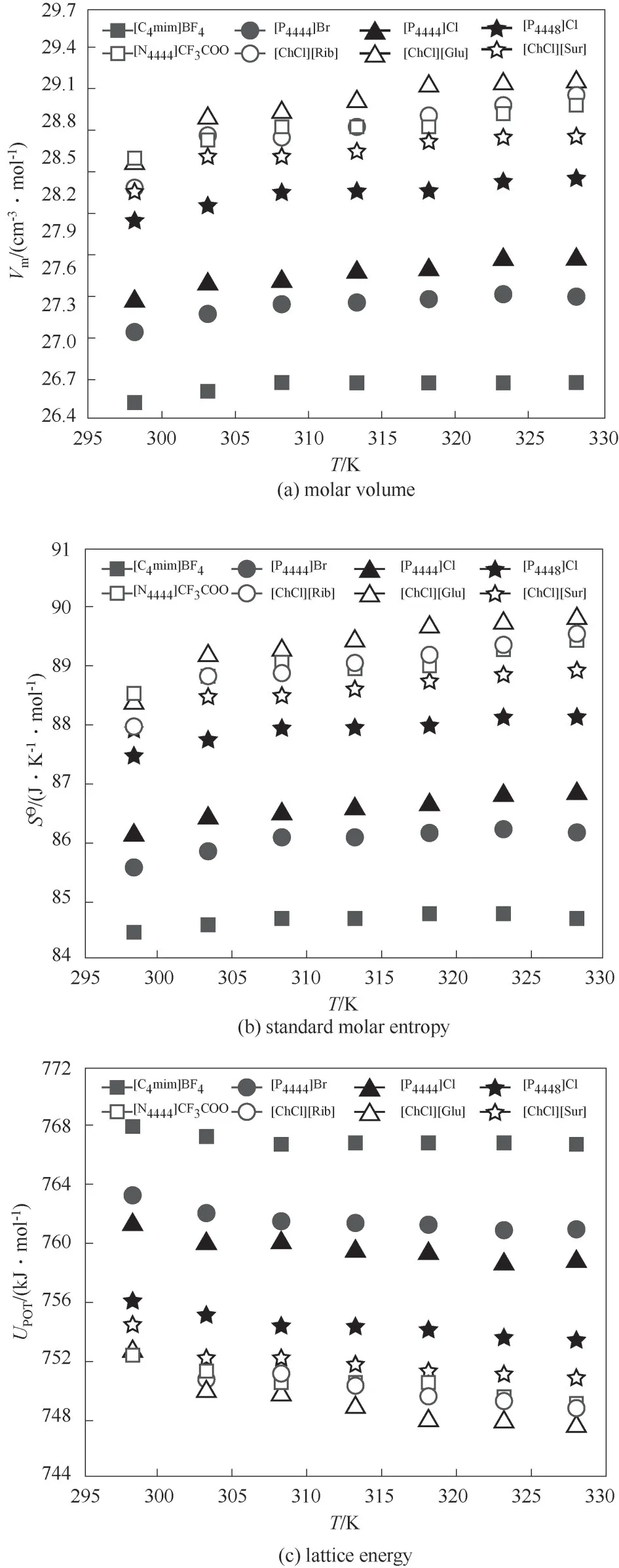
圖7 不同溫度下離子液體-低共熔溶劑-水三元體系摩爾體積(V m)、標準摩爾熵(S?)和晶格能(U POT)的變化趨勢Fig.7 Molar volume(V m),standard molar entropy(S?)and lattice energy(U POT)for ILs-DESs-H 2Oas a function of temperature
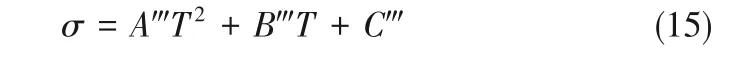
式中,σ為三元體系電導率,μS·cm-1;T為三元體系溫度,K;A?、B?、C?是擬合參數。
2.3.4 pH 為表征不同溫度下溶液中氫離子活度,測定了離子液體-低共熔溶劑-水三元體系pH隨溫度變化的趨勢,實驗數據經式(16)擬合處理。整體來看,離子液體-低共熔溶劑-水三元體系在溫度298.15~328.15 K范圍內pH呈現中性或者酸性。只有[N4444]CF3COO-[ChCl][Fru]-H2O體系呈現堿性[圖6(d)]。與離子液體-K3PO4體系或低共熔溶劑-K3PO4相比,本文構建的新型萃取體系更加溫和,利于保持生物分子的穩定性和活性。此外,相同低共熔溶劑條件下,含不同離子液體的三元體系pH規律為
[N4444]CF3COO>[P4444]Br>[C4mim]BF4>[P4448]Cl>[P4444]Cl。
由離子液體和低共熔溶劑構建的含水三元體系pH可調節范圍為3~8,受到離子液體種類影響作用明顯,可為萃取生物分子提供了設備選型和工藝設計參考。

式中,T為三元體系溫度,K;A″″、B″″是擬合參數。
2.4 離子液體-低共熔溶劑-水三元體系的量子化學計算
Hofmeister離子效應是描述特定離子對大分子在水溶液中的溶解度影響。其中,離子與溶質之間的結合最受關注。Xue等[54]應用核磁共振波譜研究了特定離子對聚(2-乙基-2-唑啉)和聚(N-異丙基丙烯酰胺)的模型化合物N,N-二甲基丙酰胺(NDA)和N-異丙基異丁酰胺(NPA)水溶液的局域環境影響,指出Hofmeister效應可能是一種全局效應,而離子與溶質的局部相互作用也具有關鍵作用。傅里葉變換紅外光譜結果表明離子對水結構的影響可能是水彎曲振動的關鍵[55]。為了探究離子液體-低共熔溶劑-水三元體系內部各分子之間的相互作用,對比分析離子液體與H2O、低共熔溶劑與H2O之間的相互作用的差別,以[P4448]Cl-[ChCl][Fru]-H2O為例,通過Gaussian 09量子化學計算軟件,得出[P4448]Cl離 子 對、[P4448]Cl-H2O(1∶1)、[ChCl][Fru]、[ChCl][Fru]-H2O(1∶1)、[P4448]Cl-[ChCl][Fru]-H2O(1∶1∶1)在DFT/B3LYP-D3/6-31G*水平優化后的最優幾何平衡構型(圖8),并計算了復合物之間的相互作用能(表4)。
當低共熔溶劑與水分子發生氫鍵相互作用時,N—Cl以及水中的H原子與Cl原子鍵長增加,氯化膽堿中的H原子與Cl原子鍵長縮短,符合“鍵級守恒原理”[56]。由于水分子的加入,[P4448]Cl-H2O體系氫鍵作用增強從而改變了P—Cl共價鍵的鍵長。單獨的Cl-電荷數為-1,當其與陽離子[P4448]+形成離子對[P4448]Cl時,電荷為-0.727。水分子使離子液體的陰離子電荷發生部分轉移,電荷的分散作用導致離子對之間的靜電作用減弱。雖然水分子與離子液體、低共熔溶劑之間都存在氫鍵作用,但是不同的二元體系內部氫鍵作用存在較大差異。例如,[P4448]Cl與H2O(1∶1)的結合能為-1046.19×103kJ·mol-1,而[ChCl][Fru]與H2O(1∶1)的結合能為-974.74×103kJ·mol-1,[ChCl][Fru]競 爭 水 分 子 的 能 力 弱 于[P4448]Cl。此外,[P4448]Cl-[ChCl][Fru]-H2O三元體系的作用能為-1972.96×103kJ·mol-1,作用強度顯著大于[P4448]Cl-H2O和[ChCl][Fru]-H2O二元體系中分子之間的作用強度。因此,離子液體、低共熔溶劑與水分子相互作用能的差異是引起雙水相體系形成的內在原因,而且這種差異越大,其形成相分離的趨勢越明顯。
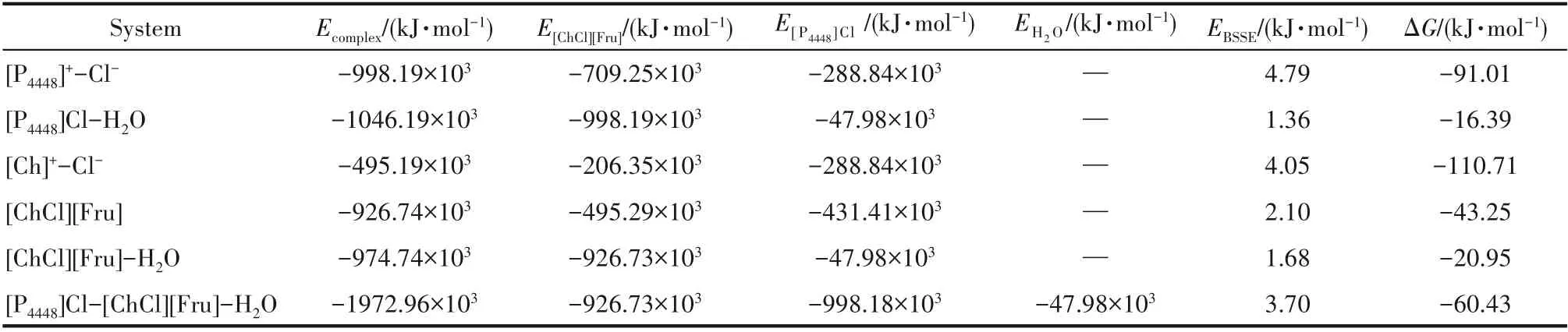
表4 離子液體-低共熔溶劑-水三元體系的相互作用能Table 4 Interaction energy of ILs-DESs-H 2O ternary system
3結 論
本文利用低共熔溶劑替代鹽類作為kosmotropic組分,以離子液體為chaotropic組分構建了具有LCST型和UCST型熱可逆相轉變行為的新型雙水相體系。離子液體、低共熔溶劑與水分子相互作用能的差異是引起雙水相體系形成的內在原因,而且這種差異越大,其形成相分離的趨勢越明顯。與低熔溶劑相比,離子液體的結構特性是決定離子液體-低共熔溶劑體系熱致相變特性和理化特性的重要因素。因此,本研究通過調控離子液體的陰、陽離子種類,與低共熔溶劑形成不同熱效應和特性的雙水相體系,可以為萃取溫敏性生物分子提供新的基礎理論和設計依據。
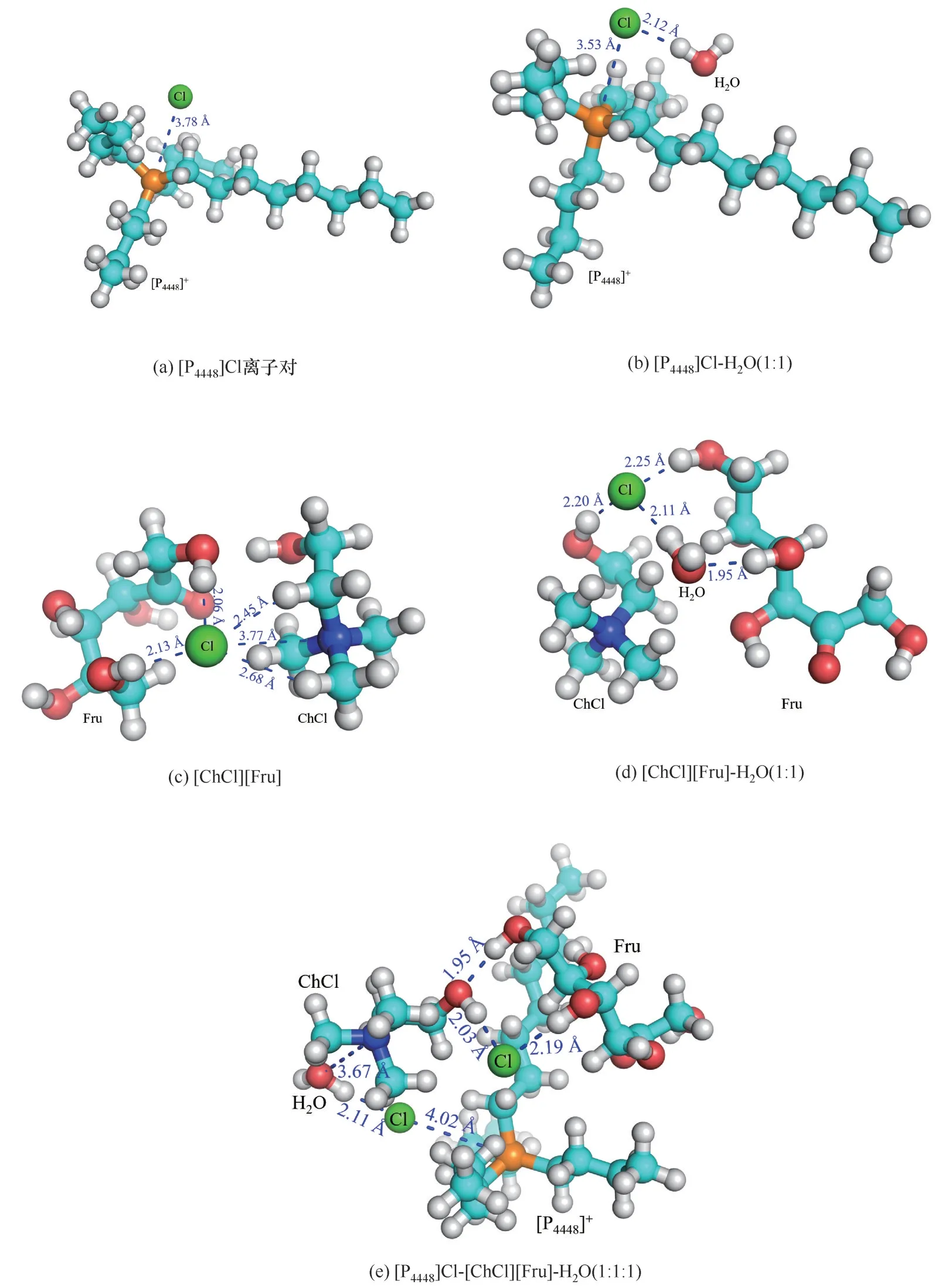
圖8 離子液體-低共熔溶劑-水三元體系幾何平衡構型(青色、藍色、紅色、綠色、橙色和白色分別為C、N、O、Cl、P和H原子)Fig.8 Geometric equilibriumconfiguration of ILs-DESs-H 2Oternary system
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
