Alport綜合征并發錐形晶狀體一例
2021-06-23 12:35:52王玉成
中國保健營養 2021年34期
王玉成
濰坊醫學院附屬醫院,山東 濰坊 261000
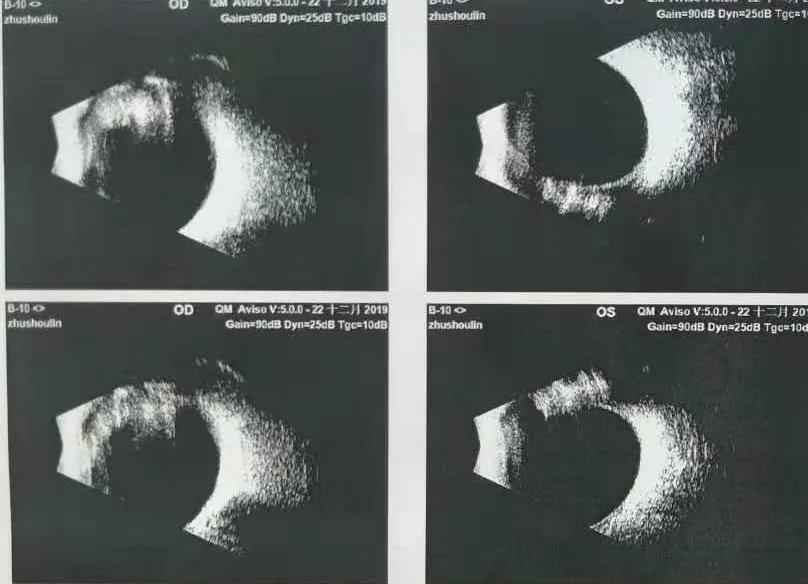
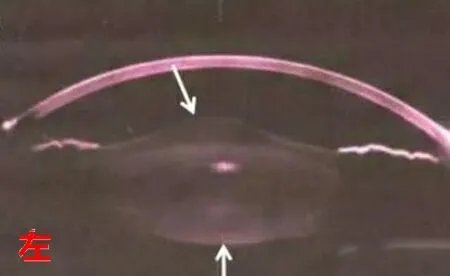
病例簡述:男,16歲。主訴:左眼視物不清2年,加重1月。現病史:2年前無明顯原因及誘因出現左眼視物不清,加重1月。既往史:2015-08-02查體檢查:尿蛋白3+,潛血3+,24小時尿蛋白:3.759g,聽力純音檢測:中度耳聾,后診斷為“先天性白內障(右)、屈光不正(雙)、弱視(右)、Alport綜合征?”。兒科擬診“腎小球腎炎”(Alport綜合征)。右眼已于外院因“先天性白內障”行白內障手術,病例記錄:視力:右眼HM/50cm(矯正無助);左眼0.3(矯正無助)。晶狀體:右眼晶狀體混濁,前囊破裂皮質溢出;左眼晶狀體混濁,鼻側圓錐狀改變。個人史:其母自訴孕早期有“上感”病史。家族史:外祖母30歲診斷為“腎炎”,治療后好轉。此次入院眼部查體:視力 右眼:0.6(-3.50DC×165=1.0)左眼:0.1(-7.5DS/-4.5DC×180=0.4) IOP:右18mmHg左17mmHg。雙眼外眼及眼前段未見明顯異常,右眼人工晶體位正,眼底未見明顯異常,左眼晶狀體圓錐狀凸起伴混濁,眼底檢查欠清。治療:2019/12/23行左眼白內障超聲乳化聯合人工晶狀體植入術,手術順利,術后恢復可,術后視力1.0,出院后囑于腎內科繼續隨診。
討 論
阿爾伯特綜合征(Alport syndrome,AS)也稱遺傳性腎炎神經性耳聾綜合征, 1875年被發現,1927年由Alport報道[1]。該病多累及腎臟、耳、眼的基底膜,從而引起結構或功能的異常。當前研究發現,AS的發病機制為COL4A5 (X-linked)、COL4A3和COL4A4(常染色體隱性遺傳)的遺傳突變,所致IV型膠原蛋白異常[2]。IV型膠原中,Heterotrimerα3α4α5(異元三聚體)在各器官的基底膜中功能中起著重要作用,如腎小球基底膜,耳蝸血管紋,角膜前、后彈力層、晶狀體囊膜,視網膜色素上皮層(Retinal Pigment Epithelium,RPE)、內叢狀層(ILM)和Bruch膜等[3]。因此當出現基因突變時,基底膜異常可引起病變發生。國外報道中,該病的流行率約為1:10000,X連鎖變異占85%,男性較女性病情重。該病的診斷需多學科綜合診斷:1、腎內科;2、耳鼻喉科;3、眼科:眼前節多有特征改變,病變嚴重時眼底檢查也會有顏色的異常。
AS在眼部表現較多見,晶狀體形態的異常,常表現為圓錐樣改變,Choi觀察Alport綜合征患者晶狀體發現前囊膜明顯變薄,各細胞器結構也明顯異常,這也證明了IV型膠原蛋白異常導致晶狀體基底膜結構異常的存在[4]。在角膜上較少表現出異常,均與IV型膠原纖維異常所致,如角膜上皮基底膜異常所致糜爛等,也可見報道并發圓錐角膜等[5]。而眼底視網膜并發癥較多,如色素紊亂、視網膜地毯樣改變、黃斑區異常暗反光等。視網膜結構中,尤其色素上皮層(RPE層)及玻璃體(Bruch膜)中,含有大量IV型膠原蛋白[6],因此,當發生異常時,視網膜結構也隨之病變、異常。
該病眼部特征性指標多采用計算顳側薄變指數(TTI),設備使用德國海德堡公司OCT測量黃斑中心凹顳內側(T1)、顳外側(T2)、鼻內側(N1)、鼻外側(N2)的視網膜厚度,公式為“TTI=(N1+N2一T1一T2)/(N1+N2)”。最佳診斷臨界值為9.47,靈敏度為73.1%,特異性100.0%。計算數值與正常兒童群體相對比,根據患兒黃斑顳側視網膜厚度是否明顯變薄得出結論。當男性患兒TTI>9.47時,可輔助診斷AS。該病確診方式還有基因檢測,對特定的基因點檢測異常時,也可確診。
眼科輔助檢查如下: