高鎂鋰比鹽湖鎂鋰分離與鋰提取技術研究進展
2021-06-30 01:27:28王琪趙有璟劉洋王云昊王敏項頊
化工學報 2021年6期
王琪,趙有璟,,劉洋,王云昊,王敏,項頊
(1北京化工大學化工資源有效利用國家重點實驗室,北京100029;2中國科學院青海鹽湖研究所,中國科學院鹽湖資源綜合高效利用重點實驗室,青海省鹽湖資源化學重點實驗室,青海西寧810008)
引 言
鋰作為元素周期表中的第一個金屬元素,是最輕的金屬,具有化學性質活潑、比熱容大、膨脹系數低等優良特性,廣泛應用于電池、玻璃陶瓷、潤滑脂、核能等領域,是現階段新能源汽車產業發展不可或缺的關鍵原料,被譽為“金屬味精”。《全國礦產資源規劃(2016—2020)》將鋰確立為我國24種戰略性礦產之一,歐盟、美國等也將鋰礦產列入其關鍵礦產目錄。
全球鋰最終用途市場如圖1所示[1]。近年來,由于可充電鋰電池在便攜式電子設備、電動工具、電動汽車和電網儲能中的廣泛應用,使得鋰在電池中的用量大幅增加,其占比從2016年的35%上升至2020年的65%。我國新能源汽車產業發展迅猛,鋰離子電池產量逐年增長,預計鋰資源的市場需求量將以每年20%的增速快速增加,對我國鋰資源的可持續供給帶來愈發嚴峻的挑戰[2-3]。
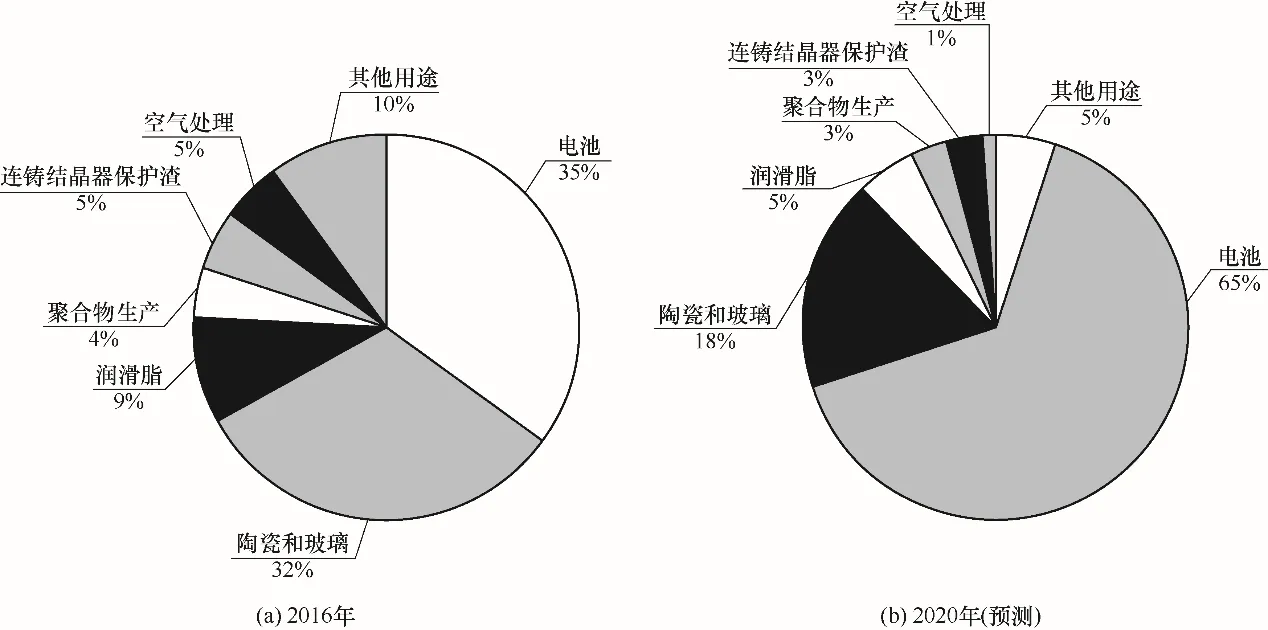
圖1 全球鋰最終用途市場分布情況[1]Fig.1 Distribution of global end-use markets of lithium[1]
根據美國地質調查局(USGS)2020年數據,世界鋰資源量約為8000萬噸,鋰儲量1400萬噸[1]。其中大陸鹵水含鋰量最為豐富,占總儲量的59%。中國已探明鋰資源量為450萬噸,其中超過71%的鋰資源蘊藏在鹽湖鹵水中,并且從鹵水生產鋰的成本通常比從硬巖源生產成本低30%~50%。我國鹽湖鋰礦主要集中分布在青藏高原,其中的鋰鹽礦主要賦存于鹽湖地表鹵水和晶間鹵水中。青海鹽湖鹵水大多屬于硫酸鎂亞型和氯化物型,青海柴達木盆地的大柴旦鹽湖、東臺吉乃爾鹽湖、西臺吉乃爾鹽湖和一里坪鹽湖是典型的硫酸鹽型高鎂鋰比鹽湖,察爾汗鹽湖鹵水屬于氯化物型,其鎂鋰比值高達1400以上。我國鹽湖鹵水具有高鎂鋰比、高鎂含量、低鋰含量的特征(表1)。鋰和鎂在周期表中呈對角線關系,物理、化學性質相近,特別是Mg2+和Li+的半徑相近,分別為72和76 pm。高鎂鋰比鹽湖鎂鋰高效分離是世界性難題[8]。
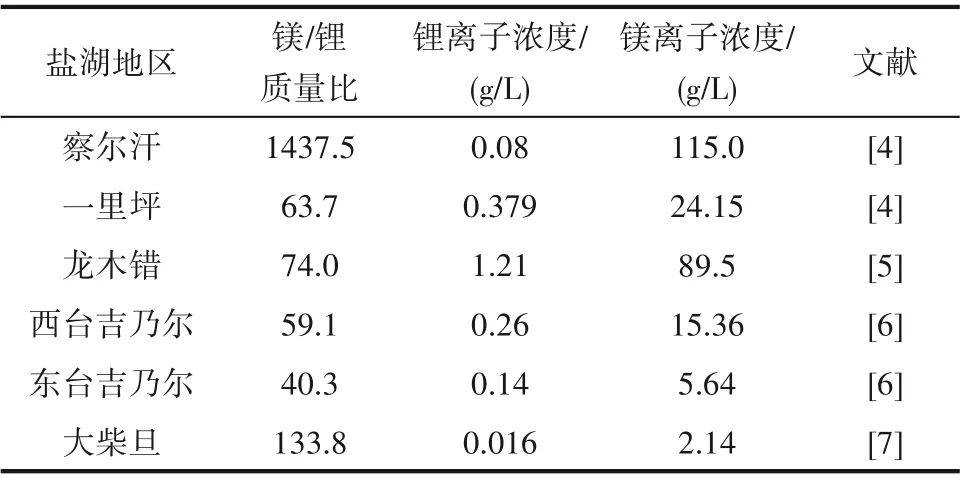
表1 中國主要高鎂鋰比鹽湖鹵水資源組成Table 1 The compositions of high Mg/Liratio salt lakebrines in China
目前全球生產的碳酸鋰原料70%來自鹽湖鹵水,僅30%來自硬巖型鋰礦。由于我國大部分鹽湖鹵水鎂鋰比值高,提取技術難度大,鋰提取率低,我國鹵水提鋰僅占20%,且鹽湖提鋰產品多為普通工業級碳酸鋰,高端產品加工原料對外依存度高,其中,進口鋰輝石加工占比66%,進口高濃度鹵水加工占比18%。一旦進口受阻,我國新能源產業、航空航天和核能等相關領域將陷入困境。因此,鹽湖鎂鋰高效分離與鋰提取技術是保障我國鹽湖鋰資源可持續利用的關鍵,是制約鹽湖資源綜合、平衡利用的“卡脖子”技術。
本文綜述了近年來鹽湖鹵水提鋰的研究進展,重點介紹了高鎂鋰比鹽湖鹵水資源的提取方法,目前高鎂鋰比鹽湖鹵水鎂鋰分離與鋰提取技術主要有:萃取法、吸附法、膜法、電化學法以及新發展的反應/分離耦合技術。詳細介紹了各種方法的特點和適用性,綜述了反應機理和應用性能,并提出了今后的發展方向。
1 萃取法
液-液萃取技術具有工藝簡單、操作條件易于控制、成本較低、萃取效率和選擇性高的優點,被認為是青海鹽湖鹵水提鋰技術中在離子選擇性及鋰收率上最為突出的一種方法,但是萃取劑的環境問題以及對萃取設備的較高要求,在一定程度上制約了該技術的產業化應用。
萃取法在20世紀60年代得到發展,利用有機溶劑對鋰的特殊萃取性能達到提取鋰的目的,其關鍵是尋找合適的萃取劑。最早用于鹽湖提鋰的萃取劑為醇或酮類。常用于鹽湖鹵水提鋰的醇、酮類萃取劑主要有丙醇、丁醇、苯甲酰三氟丙酮(HBTA)、甲基異丁基酮(MIBK)、環己酮等,其分子結構中都含有羥基或羰基等Li+配位基團[9-10]。
1.1 有機溶劑萃取
Li等[10]提出了采用β-二酮通過逆流方法從堿性鹽水中回收鋰(圖2)。HBTA-TOPO-煤油作為萃取體系,能夠有效回收鋰。萃取反應如下:
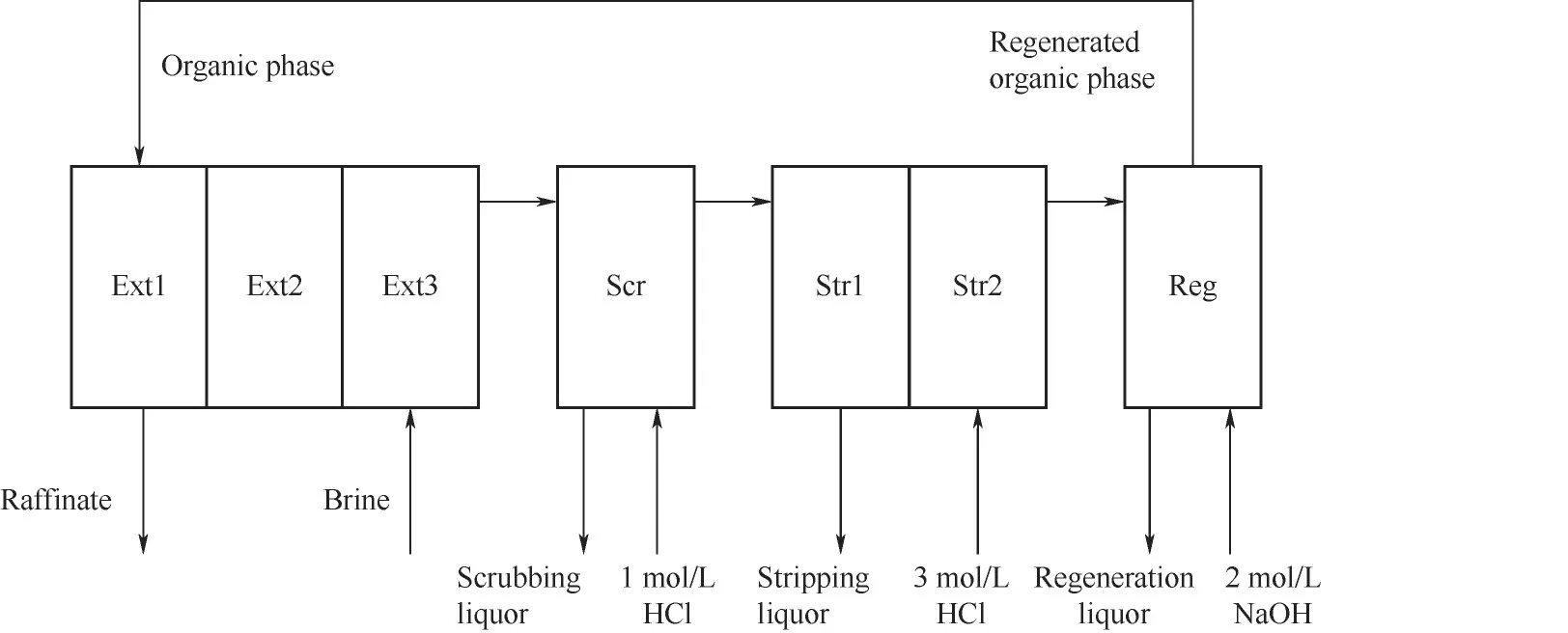
圖2 七級逆流萃取流程圖[10]Fig.2 Flow chart of seven-stage countercurrent extraction[10]
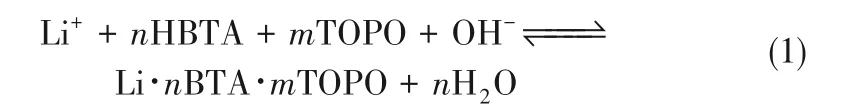
在七階段萃取過程混合沉降器中進行30 h達到穩定狀態,鋰的平均萃取率達到96%,汽提溶液中的鋰濃度為15 g/L。β-二酮類萃取劑是將含有的羥基或羰基與Li+結合為較穩定共價鍵而形成螯合結構[11]。但是,酮類萃取劑具有水溶性、易燃、易揮發等物理性質,其化學性質不穩定,限制其工業化應用[12]。
Ren等[13]模擬含高Mg/Li的鹽湖鹵水,利用磷酸三丁酯(TBP)、FeCl3和琥珀酸二乙酯分別作為萃取劑、共萃取劑和稀釋劑,進行萃取提鋰,FeCl3的添加顯著提高鋰萃取效率。萃取反應為:
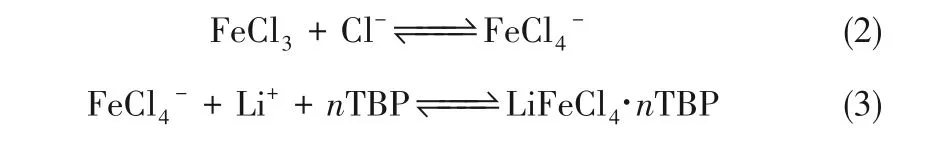
一級最高萃取效率約為65%,分離因子約為350。有機相重復利用十次,其萃取效率保持在53%。但在萃取過程中,須將FeCl3作為共萃取劑添加到有機相中,且溶液須呈酸性,在中性和堿性條件下不能進行萃取過程。
Qi等[14]提出了一種新的三元溶劑萃取體系TBP/FeCl3/P507(2-乙基己基膦酸單-2-乙基己酯),在更好地維持鋰萃取容量的同時,顯著增強了鋰的汽提過程(圖3)。85%的鋰被汽提,且Fe3+幾乎不損失。萃取反應如下:
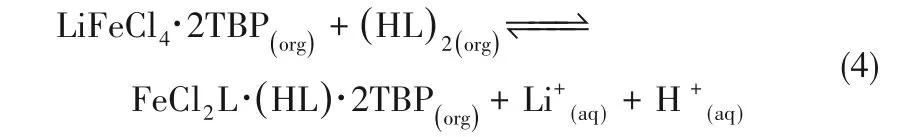
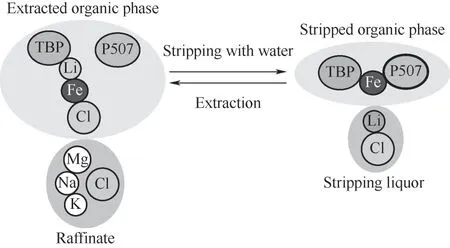
圖3 TBP/FeCl3/P507用于萃取提鋰機理[14]Fig.3 Mechanismof TBP/FeCl3/P507 for extraction of lithium[14]
他們將三元混合萃取體系TBP/FeCl3/P507用于實際鹽湖鹵水提鋰,如圖4所示通過分批和多階段模擬逆流萃取測試,發現在三階段測試中,鋰回收率達99.8%。汽提過程中,Fe3+完全保留在有機相中,使有機相可直接用于下一步提取而無須再生[15]。該體系有望可持續回收高Mg/Li+比鹽湖鹵水中的鋰。萃取反應如下:
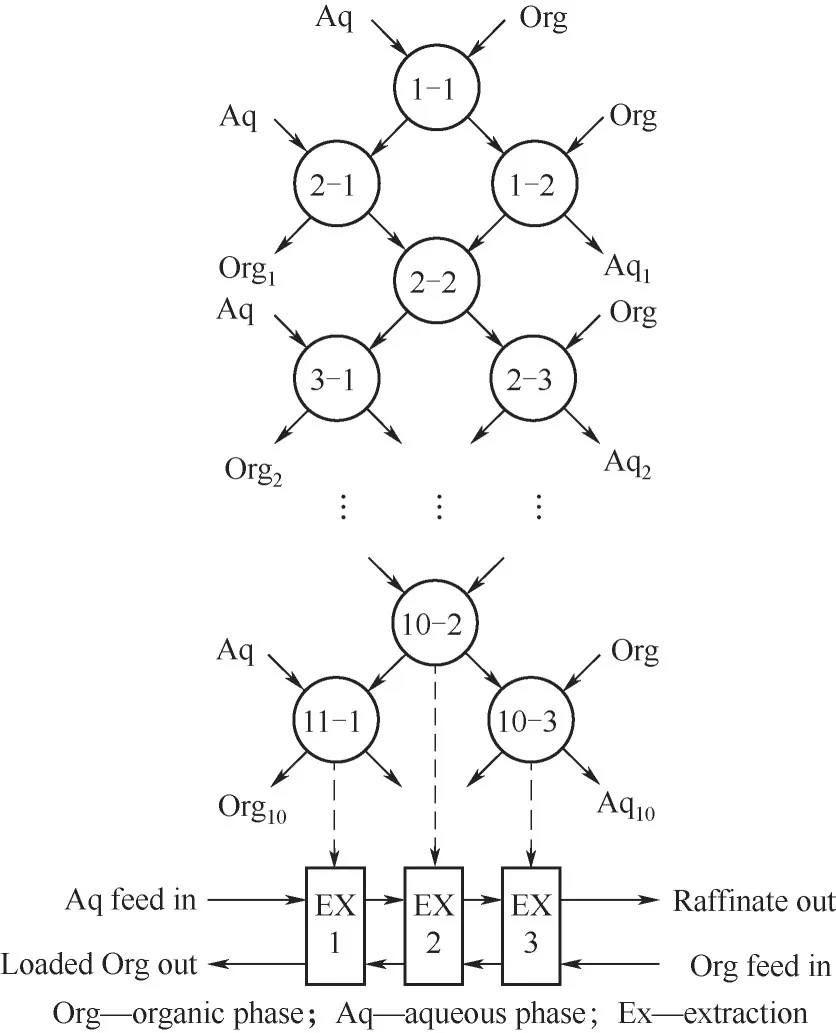
圖4 三階段SCT的過程圖[15]Fig.4 Process diagramof three-stage SCT[15]
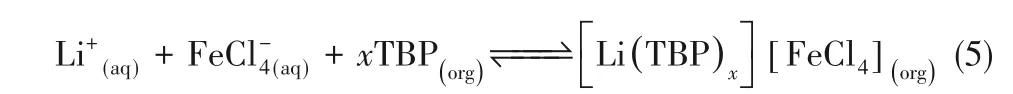
有機類萃取體系對鋰的選擇性高,目前研究更深入,也是高鎂鋰比鹽湖鹵水中具工業化應用前景的方法之一。
1.2 離子液體萃取
離子液體作為萃取分離過程的綠色溶劑,其化學性質穩定,具有不可燃性、低揮發性、可調黏度等特點。常用于鹽湖鹵水提鋰的離子液體主要有:咪唑類六氟磷酸鹽([Cnmim][PF6])、咪唑類雙(三氟甲基磺酰)酰亞胺[Cnmim][NTf2]、四丁基銨雙(2-乙基己基)-磷酸鹽[N4444][DEHP]、雙(2-乙基己基)-磷酸四辛基銨[N8888][DEHP]等[16-18]。
Deng等[19]研究了磷酸三異丁酯(TIBP)和幾種離子液體(PF6-和NT作為陰離子)對Li+萃取性能的影響。在TIBP-ILs-煤油這一萃取體系中,這些離子液體的陰離子用作共萃取試劑,具有良好的疏水性和類似于FeCl4-的立體對稱結構。以1-丁基-3-甲基咪唑六氟磷酸鹽作為離子液體展現出更好的鋰離子萃取效率,在最優條件下,單步萃取效率達83.71%,萃取反應如下:

Jia等[20]利用離子液體[C4mim][NTf2]和中性的TBP作為萃取劑對含鎂離子的鹽湖鹵水進行了鋰的萃取。在最優的條件下,鋰的萃取效率為92.37%。在萃取過程中,通過陽離子Li+和[C4mim+]之間的交換機理,將鋰離子萃取到有機相中,證明這一萃取體系能夠有效提取鹽湖鹵水鋰。
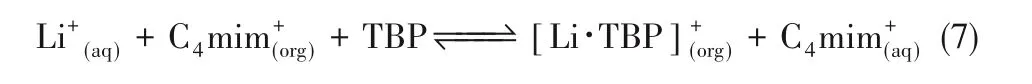
Jia等[18]合成了兩種非氟化的功能化離子液體(ILs)萃取劑,即[N4444][DEHP]和[N8888][DEHP],對溶液中的鋰進行了萃取。在溫和條件下,無須在溶液中加入復雜的絡合劑,便能將鋰完全汽提,且在循環過程中,該萃取劑的萃取效率不降低。萃取反應如式(8):

Ren等[21]利用雜多酸離子液體回收鋰離子,合成了無氟離子液體1-丁基-3-甲基咪唑磷鎢酸鹽([Bmim]3PW12O40),將其作為共萃取劑從高Mg/Li比鹽湖鹵水中回收鋰。在最佳條件下,通過五級錯流萃取,Li+總萃取率為99.23%。高選擇性萃取鋰離子是通過TBP與Li+之間的陽離子交換和配位作用實現的。10個再生循環萃取過程表明,有機相對Li+的選擇性萃取具有高的穩定性。
深共熔溶劑被認為是一種新型的離子液體類似物,它們通過氫鍵相互作用由兩組分或三組分共晶混合物自締合形成,由于低揮發性、低成本可生物降解性和低毒性,被認為是綠色溶劑。Zhu等[22]利用一種疏水性深共熔溶劑(HDES)提取鋰離子,綠色協同系統TBP-HDES具有高選擇性鋰萃取能力,單次萃取率達到76.8%,5次循環后萃取率仍能達到60%。
離子液體作為可替代有機萃取劑的綠色體系,雖然拓寬了萃取法的應用范圍,降低了污染,但價格偏高,一定程度上制約了其工業應用。
2 吸附法
吸附法是一種從環境和經濟角度具有較大優勢的鋰提取技術。尤其是從低品位的高鎂鋰比鹵水、海水中提鋰的優勢更明顯。該方法的關鍵在于發展性能優異的吸附劑,從最初的有機吸附劑到無機離子交換劑,吸附法已實現從稀溶液中提鋰。現階段吸附劑主要分錳系、鈦系離子篩和鋁系吸附劑。錳系離子篩主要為尖晶石結構的鋰錳氧化物離子篩,通過復合吸附機理進行鋰的選擇性吸附;鈦系離子篩主要為層狀結構與尖晶石結構的鋰鈦氧化物,通過離子交換進行選擇性吸附鋰離子;鋁系吸附劑主要是LiCl·2Al(OH)3·nH2O,利用有序結構空位進行選擇性吸附鋰。
2.1 錳系離子篩
通過將鋰離子引入錳化合物中,熱處理形成尖晶石結構,再利用酸處理用質子置換Li+,在不改變晶體結構的情況下,形成錳系鋰離子篩(圖5)。在多離子共存下,鋰離子篩具有篩選和記憶目標鋰離子的能力,稱為“離子篩效應”[23]。
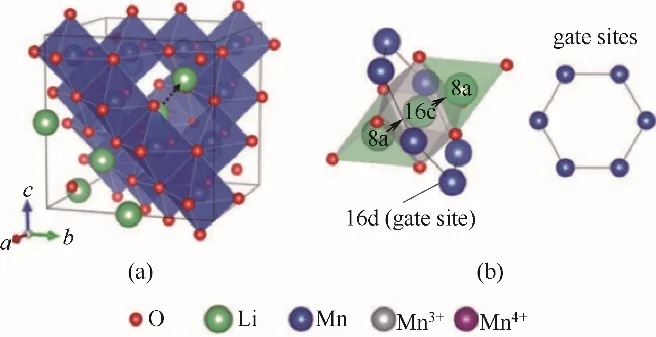
圖5 尖晶石LiMn2O4的晶體結構(錳離子駐留在氧離子形成的八面體中。虛線箭頭表示鋰的擴散路徑)(a);鋰擴散通道從四面體8a位置通過由六個空位包圍的八面體16c空位到相鄰的8a位置的示意圖(b)[23]Fig.5 Crystal structure of spinel LiMn2O4(Manganese ions reside in the octahedra formed by oxygen ions.The dotted arrow denotes a lithiumdiffusion path)(a);Schematic illustration of the lithium diffusion channel from a tetrahedral 8a site to an adjacent 8a site through an octahedral 16c vacancy surroundedby six manganeseions in the octahedral 16d gate sites(b)[23]
目前,對錳系鋰離子篩吸附機理有三種解釋。

(3)復合反應機制:吸附過程既涉及氧化還原機制,也涉及離子交換機制[27]。
LiMn2O4、Li1.6Mn1.6O4和Li4Mn5O12等需要通過酸處理形成錳系離子篩(LMOs)。由于Mn3+的特殊電子結構,在酸性環境中引起LMO晶格結構嚴重畸變,體相中Mn3+的電子被轉移到Mn4+,Mn4+被還原為Mn2+后溶解[28]。LMO的錳溶損阻礙了錳系離子篩的工業應用。
錳系離子篩λ-MnO2、MnO2·0.31H2O和MnO2·0.5H2O 分 別 由 前 體 LiMn2O4、Li1.33Mnl.67O4和Li1.6Mn1.6O4形成,理論吸附容量分別為5.7 mmol/g(39.6 mg/g)、8.5 mmol/g(59.0 mg/g)和10.5 mmol/g(72.9 mg/g)[29]。盡管理論容量相對較高,但由于LMO穩定性差和錳溶解速率高,實際吸附容量相對較低。改性LMO可提高吸附能力,降低錳的溶解損失。常見方法為摻雜過渡金屬,如銅、鋅、鐵、鎳和鈦修飾LMO。金屬與氧原子之間可形成強離子鍵,Li-O鍵長度增加,Mn-O鍵長度減小,LMO尖晶石結構的晶格收縮,增強結構穩定性,阻礙錳溶解。
鎂摻雜對增強LMOs結構穩定性具有積極的影響。Zhao等[30]采用軟化學方法制備LiMg0.56Mn1.50O4,經過五次循環后,鋰提取率仍大于95%。通過摻雜鐵也可降低錳損失,利用鐵的磁性有利于回收吸附劑。
由于鋰離子篩呈粉末狀,在工業應用中流動性差、滲透性不足、循環效率低,柱運行時壓降大、粉體損失大、能耗高,工業生產中對離子篩進行如造粒、成膜、發泡、成纖維、磁化等成型處理。Yu等[31]合成LiMn2O4、Li1.66Mn1.66O4和Li4Mn5O12等鋰錳氧化物(LMO),應用于地熱水中鋰選擇性回收,發現粉狀Li4Mn5O12對低濃度Li具有較高吸附性能,在殼聚糖(CTS)/LMO質量比3∶2下造粒(圖6),在303.15 K時,地熱水中Li平衡吸附容量達到8.98 mg/g,Li與其他離子的分離系數均高于5,經過五次吸附-解吸循環后,吸附容量衰減不超過1.1%[31]。
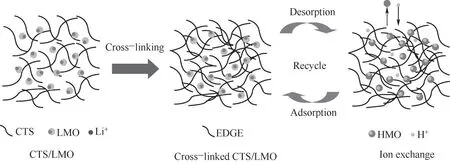
圖6 用EGDE交聯球形CTS/LMO示意圖[31]Fig.6 Schematic diagram of cross-linking spherical CTS/LMOwith EGDE[31]
Ju等[32]報道了通過浸漬結晶和鈣化方法后將鋰錳氧化物(Li4Mn5O12)負載到管狀α-Al2O3陶瓷襯底上,作為離子篩前體,酸洗后得到鋰離子篩。在動態吸附-解吸過程,該鋰離子篩對Li+吸附容量為9.74 mg/g,錳溶損為0.99%。經過3次循環,該吸附劑保持80%的初始動態吸附容量。
錳系鋰離子篩對Li+具有良好的選擇性,可從高鎂鋰比鹵水中吸附Li,但在解吸過程中,錳溶解阻礙了其工業應用。
2.2 鈦系離子篩
鋰鈦氧化物(LTOs)離子篩對鋰具有較高的選擇性,該吸附劑可在其晶體結構無重大變化的條件下,實現鋰離子的插層和脫出,而鹽水中存在的其他離子如Na+、K+和Ca2+,由于半徑較大,不被吸附,鈦系離子篩的離子選擇性順序為Li+?Na+>Mg2+>Ca2+>K+。LTOs如Li2TiO3和Li4Ti5O12可通過鋰鹽處理商業二氧化鈦來合成,在洗脫鋰時表現出更好的穩定性。
在Li2TiO3的晶體結構中,氧原子排列在立方體晶格位點,鋰和鈦占據八面體空隙,形成層狀單斜結構,一層包含Li原子,另一層包含LiTi2序列(圖7)[33]。用酸將鋰從Li2TiO3中洗脫出來,得到H2TiO3,用于吸附鋰[式(11)~式(13)]。在鋰吸附/解吸過程中,LTOs具有較好的結構穩定性,可回收利用。但在酸浸過程中,Li2TiO3會發生結構重排,特別是在高濃度氯離子存在下,會導致其轉化為金紅石相TiO2,對Li的吸附能力降低[34]。

圖7 鋰鈦酸鹽的晶體結構[33]Fig.7 Crystal structure of lithiumtitanate[33]
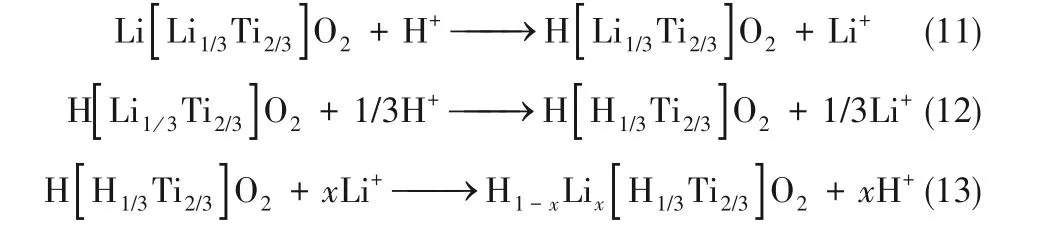
Tang等[35]采用靜電紡絲和煅燒相結合的方法制備了多孔鈦基鋰離子篩(LIS)納米纖維,具有良好鋰吸附容量和較高選擇性,如圖8所示。焙燒形成的多孔結構增加了吸附位點暴露,顯著加速了鋰在骨架空位的脫出與嵌入。該材料可在30 min內達到吸附平衡,吸附容量59.1 mg/g,接近理論值(63.77 mg/g)。具有良好的穩定性,循環6次后對鋰離子吸附量仍為86.5%。
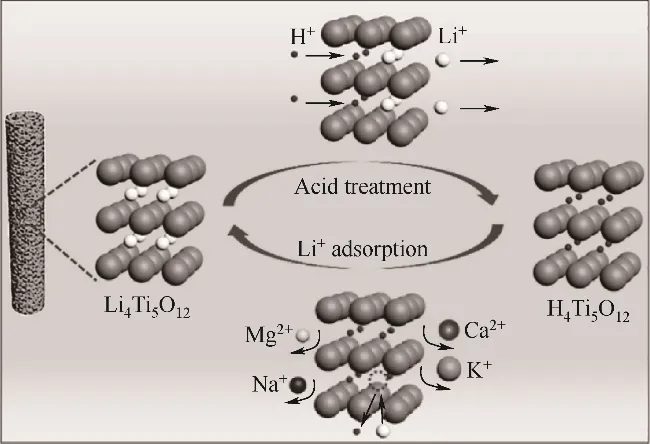
圖8 P-HTO-NF鋰回收示意圖[35]Fig.8 Schematic diagram of P-HTO-NFlithium recovery[35]
Li等[36]發現Li2TiO3可以實現鹽湖鹵水中鎂/鋰分離,Li/Mg分離系數為4783。He等[37]發現Li2TiO3脫鋰后形成的H2TiO3吸附容量達到57.8 mg/g,在5次循環后,吸附容量為25~30 mg/g。雖然Li2TiO3具有較高的理論吸附容量(142.9 mg/g),但由于實際生成物中鋰含量低于理論量,以及合成Li2TiO3時的高溫條件引起顆粒間團聚,所得到的吸附容量僅為理論值的40%[38]。Zhu等[39]用不同TiO2前體制備鋰離子篩,發現了鋰離子篩親水性與吸附性能之間的相關性,其中銳鈦礦TiO2衍生的鋰離子篩具有更強的親水性,表現出更高的吸附性能,24 h吸附容量達到34.2 mg/g,對Li+具有很高的選擇性,且易于再生。另外,鐵摻雜的Li2TiO3離子篩Fe/Ti-0.15(H)具有良好的大規模連續磁回收率(96%)和優良的鋰吸附容量(53 mg/g),可與懸浮液有效分離[40]。
2.3 鋁系吸附劑
鋁系吸附劑一般化學式為LiCl·2Al(OH)3·nH2O,是將LiCl插入Al(OH)3中形成鋰鋁復合金屬氫氧化物(LiAl-LDH)而產生的一種有序空位型層狀結構[圖9,式(14)][41],是目前我國青海鹽湖唯一大規模工業化應用的吸附劑。

圖9 Al(OH)3吸附Li+轉化為LiAl-LDH示意圖[41]Fig.9 Schematic diagramof Al(OH)3 adsorption of Li+into LiAl-LDH[41]
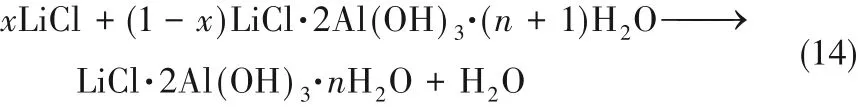
因為Li+可以進入Al(OH)3晶格,占據八面體空穴,而體積較大的堿金屬和堿土金屬離子由于空間效應不能進入,使得鋁系吸附劑對Li+具有選擇性。雖然Mg2+的離子半徑(72 pm)與Li+的離子半徑(76 pm)相似,但Mg2+與水分子結合形成水合離子([Mg(H2O)6]2+),離子半徑增大到428 pm,而Li+只能形成半徑382 pm的水合離子,水化自由能遠小于Mg2+(Li+為515 kJ/mol,Mg2+為1922 kJ/mol)[42-46]。因此,實現鋁吸附劑對Li+的吸附和鎂鋰分離。
Yu等[47-49]研究了具有結構缺陷的LiCl·2Al(OH)3·nH2O吸附劑,從高Mg/Li比鹽水中提取鋰,吸附容量為7~8 mg/g,鹵水中含有高濃度的MgCl2,吸附劑對鋰仍具有選擇性[47]。當MgCl2濃度從0增加到500 g/L時,鋰吸附容量提高近4倍[48]。進一步研究了吸附劑對Li-Na-MgCl2體系的協同競爭吸附,驗證了鋰和鈉離子之間的競爭吸附作用,說明了在柱吸附過程中回收鋰的可能性,為柱吸附實驗提供了數據[49]。
Yu等[50-52]用超順磁性納米粒子(Fe3O4或γ-Fe2O3)摻雜入鋁吸附劑,無須造粒過程,并實現外部磁場下的吸附劑回收。用磁性Fe3O4化學共沉淀法制備磁性Li/Al-LDH(MLDH),用磁棒進行吸附劑與溶液分離。發現MLDHs的鋰吸附容量下降,而選擇性增大。當Fe3O4含量為13.11%(質量)時,MLDH對鋰吸附容量為5.83 mg/g,吸附劑回收率為97%。在固定床柱中填充成型吸附劑,模擬從鹽水中吸附鋰離子。發現初始鋰濃度、床層高度和進料流量對柱吸附性能有顯著影響[51]。為了模擬和預測鋰的吸附曲線,建立了均勻表面擴散模型[52]。雖然鋁系吸附劑已工業應用,但吸附容量低,需要進一步優化。
吸附法適用于高Mg/Li比鹽湖鹵水提鋰工藝。鋰吸附劑具有優異的鋰選擇性、較高的理論鋰吸附容量和優越的循環性能,并且可從較低鋰濃度的溶液中有效提取鋰,因此該技術未來在鹽湖原始鹵水提鋰以及西藏鹽湖鋰資源開發中具有較大的應用潛力。但現階段,鋰吸附劑還有諸多問題及挑戰:
(1)實際吸附容量與理論吸附容量之間存在巨大差距,這可能是由于吸附劑前體洗脫過程中Li+脫附不完全以及循環過程中吸附通道被堵塞,導致有效空位數減少;
(2)在洗脫過程中,粉末吸附劑的核心骨架在溶液中溶損、破裂、塌陷;
(3)工業生產中對吸附劑進行如造粒、成膜、發泡、成纖維、磁化等處理,導致吸附位點被堵塞和覆蓋,使工業吸附劑的吸附容量大幅降低。因此,后續探究優良的造粒成型方法提高吸附容量是吸附劑工業應用的關鍵。
3 反應/分離耦合技術
反應/分離耦合是指能同時實現化學反應和物理分離,其主要特點是:
(1)在反應過程中分離出具有抑制作用的產物,可提高總收率和處理能力;
(2)在反應過程中消除不良物質,從而保持較高的反應速率;
(3)反應過程中產生的熱量促進分離過程,從而降低能耗;
(4)簡化后續分離過程,從而降低生產成本。
該技術在反應精餾、反應-膜分離和生物反應/分離中有廣泛應用[53-57]。針對我國高鎂鋰比鹽湖鎂鋰分離的重大難題,Guo等[58]提出反應/分離耦合創新思想,依據層狀復合金屬氫氧化物(layered double hydroxides,LDHs)的晶格選擇性與離子識別科學原理,在鎂鋰分離、提取鋰的同時聯產鎂基功能材料,例如:MgAl-LDHs。MgAl-LDHs層板由MgO6和AlO6八面體交替排列組成,當鎂離子和鋰離子在溶液里同時存在時,Mg2+會形成穩定的金屬-氧八面體結構即MgO6,而Li+不能形成八面體結構,即不能進入LDHs的層板,使得MgAl-LDHs對Mg2+具有晶格選擇性。因此,鹵水中的Mg2+與外加Al3+在堿作用下反應,形成MgAl-LDHs固體,鎂離子選擇性進入固相形成MgAl-LDHs,而鋰離子仍保留在液相,實現鎂鋰高效分離。在高效回收鋰的同時,充分利用了鹽湖豐富的鎂資源(圖10)。鎂基功能材料在紫外阻隔、氣體阻隔、PVC熱穩定劑、阻燃/抑煙、催化、土壤修復治理等領域應用廣泛。
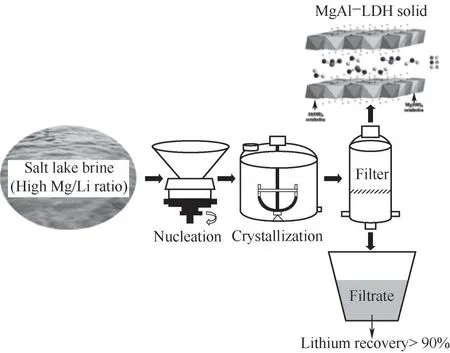
圖10 反應/分離耦合技術從鹽湖鹵水中分離鎂、鋰并制備LDHs產品流程[58]Fig.10 Scheme of Mg/Liseparation and LDHs preparation by reaction-coupled separation technology[58]
Xiang等[59]從實驗和理論模擬兩方面提出了混合離子溶液中Mg/Li分離的條件判據(圖11),為反應/分離耦合技術應用于鹽湖鎂鋰分離提供了理論依據。確定了鎂離子和鋰離子分離的邊界條件是Mg/Al摩爾比大于2,而與溶液中的Mg/Li質量比無關。利用微液膜反應器進行分離過程強化,通過研究成核和結晶過程條件,以鋰損失、反應后Mg/Li比和LDHs產品粒徑為主要指標,發現當形成高結晶度的LDHs時,顆粒間團聚被顯著抑制,LDHs產品粒徑更均一,反應/分離過程的鋰損失降低。反應/分離后溶液中的鎂鋰比由初始的12.66大幅降至0.08,鋰回收率高于90%。
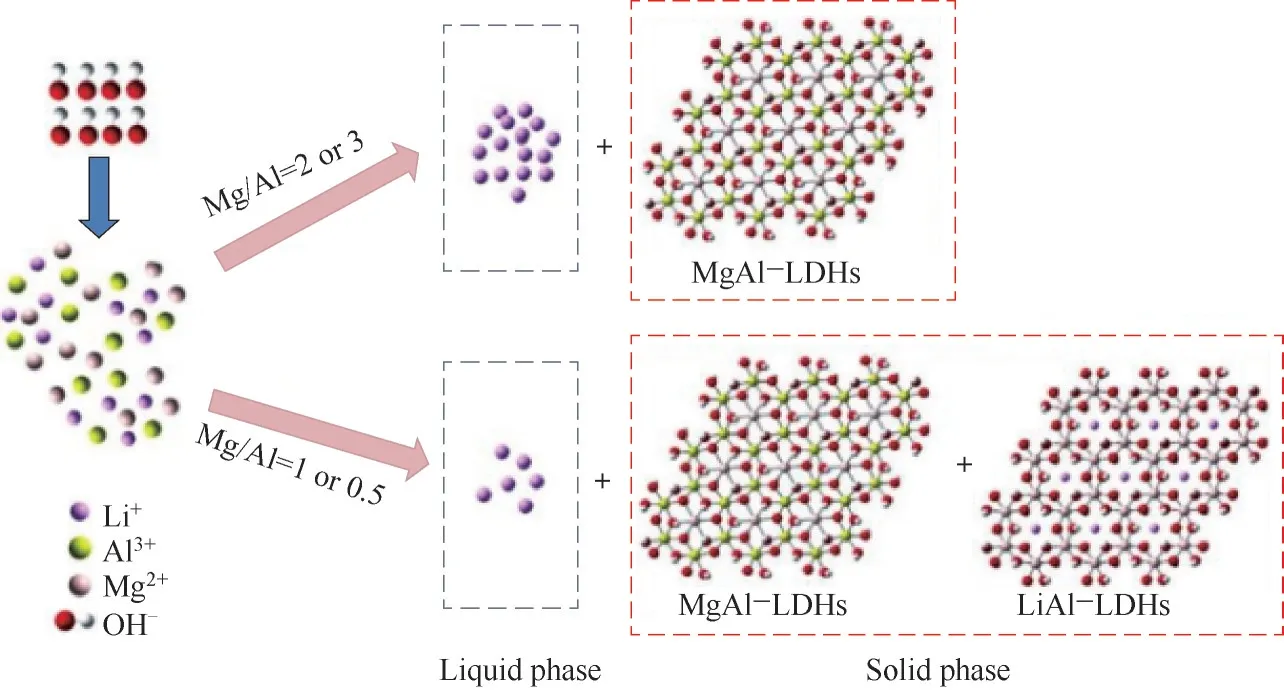
圖11 反應/分離耦合技術分離鹽湖鹵水中鎂、鋰的理論判據邊界條件[59]Fig.11 Theoretical criterion and boundary conditions for separation of magnesium and lithium in salt lake brine by reaction-coupled separation technology[59]
在利用反應/分離耦合技術實現鹽湖鎂、鋰分離時,由于向鹵水溶液引入了Na+,除鎂后溶液的Na/Li比高達48.7,這為鋰的提取帶來了新挑戰。依據晶體結構分析,鋰離子可以進入Al(OH)3結構的有序空位,形成穩定的空位嵌入型鋰鋁氫氧化物(LiAl-LDHs)(圖12)[60]。因此,Xiang等[61]進一步采用反應/分離耦合技術從高鈉鹵水中提取鋰,利用LiAl-LDHs的離子識別結構特征,從提鎂后的高鈉鹵水中選擇性地捕獲鋰離子,使鋰離子進入固相,而鈉離子則保留在液相,實現了高效鋰、鈉分離。通過脫鋰反應將LiAl-LDHs中的鋰離子脫出,轉變為鋰空位型Al(OH)3,可從鹽湖鹵水中選擇性地捕獲鋰離子,實現高選擇性鋰提取。該研究實現了從預合成的LiAl-LDHs中脫鋰、從鹵水中提鋰的循環過程。在吸附過程中,鋰離子被捕獲進入AlO6形成的晶格空位中,而Cl-插入到層間,恢復為LiAl-LDHs結構,吸附容量達到22.94 mg/g,鋰回收率達到96%。反應/分離耦合技術應用于高鈉鹵水提取鋰,證實了LiAl-LDHs在選擇性捕獲鋰離子中的關鍵作用,拓寬了該技術的適用范圍。
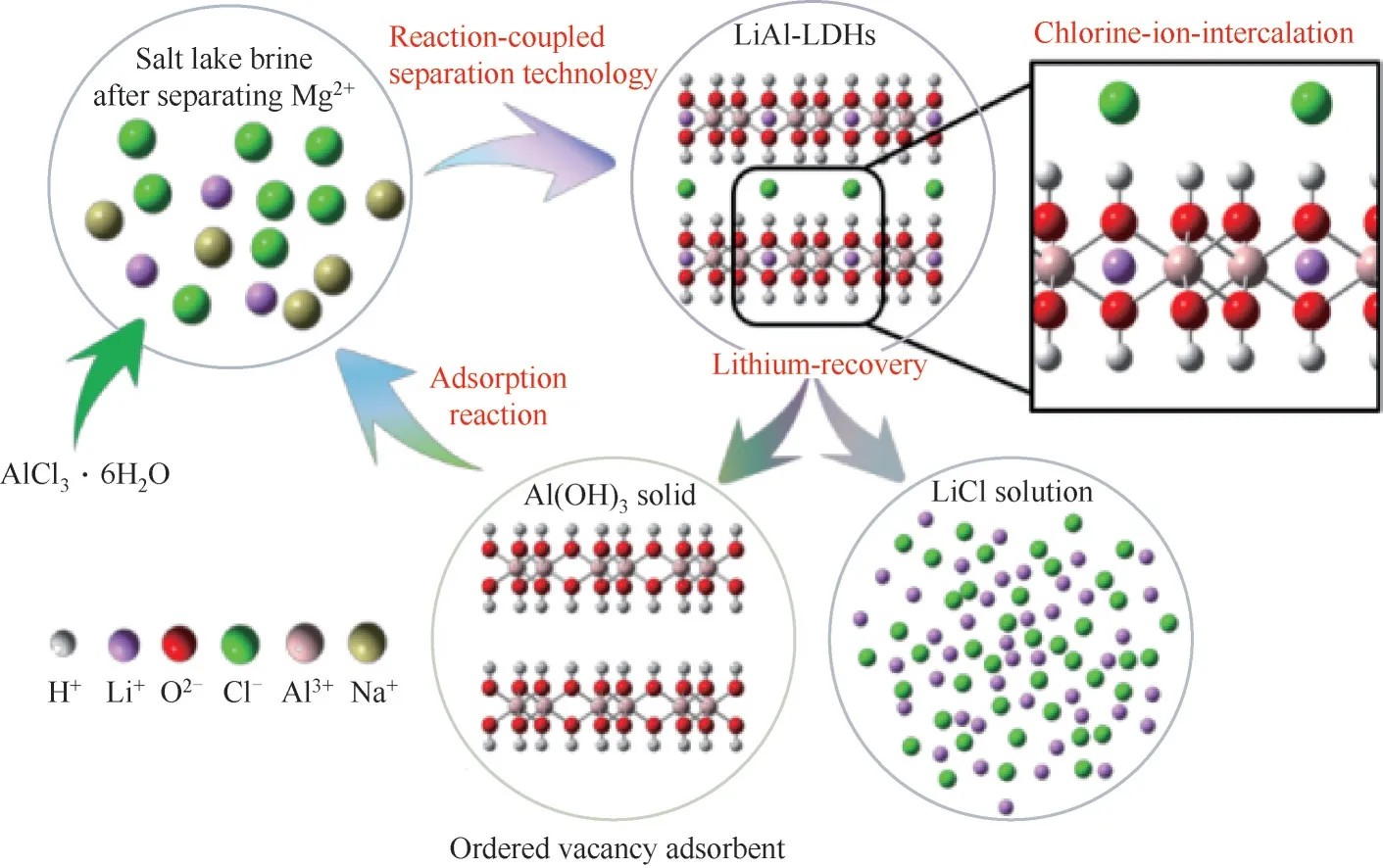
圖12 合成LiAl-LDHs并制備有序空位型鋰吸附劑高選擇性捕獲鋰離子[60]Fig.12 Scheme of selective capture of lithium ions by ordered vacancy adsorbent prepared by LiAl-LDHs[60]
反應/分離耦合技術打通了從鹽湖鹵水鋰資源提取到鋰鹽富集的整個技術鏈條,通過鋰回收和吸附反應中晶相的改變實現了循環過程,反應條件溫和,是鹽湖鋰資源提取利用新工藝的有益探索。該技術已在青海察爾汗鹽湖提鋰項目中完成中試,為后續我國青海其他鹽湖及西藏鹽湖提鋰提供了新的技術方向。
4膜 法
膜法是一種具有較高分離效率的新興技術,且膜過程已實現規模化連續運行以及自動化控制,在鹽湖鹵水鋰資源提取領域具有廣闊的應用前景。根據驅動力不同,膜法提鋰技術主要分為納濾法(壓力驅動)與離子選擇性電滲析法(電位驅動)。此外,相比于傳統電滲析,雙極膜電滲析和膜電容去離子系統在工藝和功能上有了較大改進,因此受到了關注。近年來,許多膜材料相繼被開發并用于鹽湖鹵水鋰的分離與提取,具有高選擇性、低能耗和良好循環性能的膜材料是膜過程工業應用的關鍵。
4.1 納濾
納濾是一種壓力驅動的膜分離過程,受Donnan效應和尺寸篩選效應的影響,對多價離子具有更好的保留性能,對單價離子具有更好的滲透性[62]。然而,在實際操作中,納濾膜并不能完全分離鎂和鋰,只可使鹽湖鹵水中Mg/Li比明顯降低,從而降低后續鋰提取過程中的分離難度。在納濾分離過程中常出現膜污染現象,而且延長操作時間后,分離效率降低。因此,該技術在實際應用中應與其他分離方法相結合,以提高分離效率、延長使用壽命、降低分離成本[63]。
采用模擬高Mg/Li比鹵水溶液研究DK納濾膜,發現其對鎂和鋰具有良好的分離效果。Wang等[64]通過提高操作壓力,增加了鋰收率,改善了鎂鋰分離效果,在3.5 MPa下觀察到鎂截留率92%。他們還研究了pH對鎂鋰分離的影響,在較低pH時,由于多價離子的介電排斥增強,Li+和Mg2+的分離得到改善[65]。在兩階段納濾過程中,溶液pH是至關重要的,在pH=3.5時,分離后Mg2+/Li+比從13.25下降到0.17。Yu等[6]詳細研究了操作時間、操作壓力、進料溫度、pH、鎂鋰比等對DL-2540NF分離鎂鋰的影響,提高操作壓力,降低進料溫度和pH,可提高Mg2+截留率,有利于鎂鋰分離,同時Mg2+/Li+比增大、競爭性單價離子(如Na+和K+)的存在會增加Li+截留率,不利于鎂鋰分離。
Zhang等[66]將三丁磷酸(TBP)/FeCl3經典萃取體系加入到聚氯乙烯(PVC)基聚合物包裹膜(PIM)中,從富含Mg2+的溶液中提取Li+(初始Mg2+/Li+摩爾比為15)。與液-液萃取相比,聚合物包覆膜對Li+提取率提高了20%,Li+與Mg2+分離系數提高了5%,為鹵水中提取鋰提供了一種有潛力的膜技術。
Li等[67]合成了用于鎂/鋰分離的聚酰胺復合納濾 膜,對 離 子 的 截 留 順 序 為MgCl2>MgSO4>NaCl>LiCl。當MgCl2濃度為2 g/L、初始Mg/Li比為20時,通過納濾過程可將Mg/Li比降至7.7。當等電點小于9.5時,膜表面帶正電(商用NF90膜的等電點為3.6)。因此,合成納濾膜的性能優于商用NF90。Li等[68]用EDTA改性納濾膜,表面帶正電荷,可與二價陽離子如Mg2+形成配合物,鎂和鋰的分離因子為9.2。Xu等[69]制備了一種含羥基多壁碳納米管修飾的納濾膜,處理鹵水的鎂鋰質量比為14,鎂和鋰的分離因子為10.4,具有97%以上的二價離子截留率和高滲透性。Peng等[70]構建了一種新型聚合物功能化金屬-有機骨架(MOF)納濾膜,具有優異的鋰離子選擇性,分離因子高達1815,可實現極快鋰分離。Zhao等[71]使用含二齒胺基團的電解質單體(DAIB)改性原始聚酰胺復合膜,DAIB改性不僅提高了膜表面親水性,而且還降低了水通過100 nm厚度分離層的阻力,表現出高透水性,在200 h連續納濾中,穩定性良好。
在納濾膜材料結構設計方面,首先應開發鋰離子通量較高的納濾膜,以降低分離成本;提升納濾膜的耐污染性和分離穩定性,因為在復雜鹵水體系下長期運行易形成膜污染,進而降低水通量和離子選擇性,不利于提升鋰回收率和鎂鋰分離效果;開發高性能的膜,提升納濾膜對多價陽離子的分離效果,這對于鎂和鋰的分離尤為關鍵。
4.2 電滲析
在電滲析過程中,溶液中的離子在電場下通過膜遷移,這種現象廣泛應用于鹽水淡化、工業廢水處理和有機酸生產等領域[72]。使用交替放置的陽離子和陰離子交換膜的電滲析過程示意圖見圖13,陽離子在電場作用下通過陽離子交換膜,而陰離子則通過陰離子交換膜遷移到電極上[73]。電滲析分離離子的原理是單價陽離子(例如:Li+、Na+、K+)通過單價選擇性陽離子交換膜遷移到濃縮室,而二價陽離子(例如:Ca2+、Mg2+)被阻擋,留在脫鹽室。選擇性離子交換膜是電滲析技術應用于鹽湖鹵水中提取鋰的關鍵。
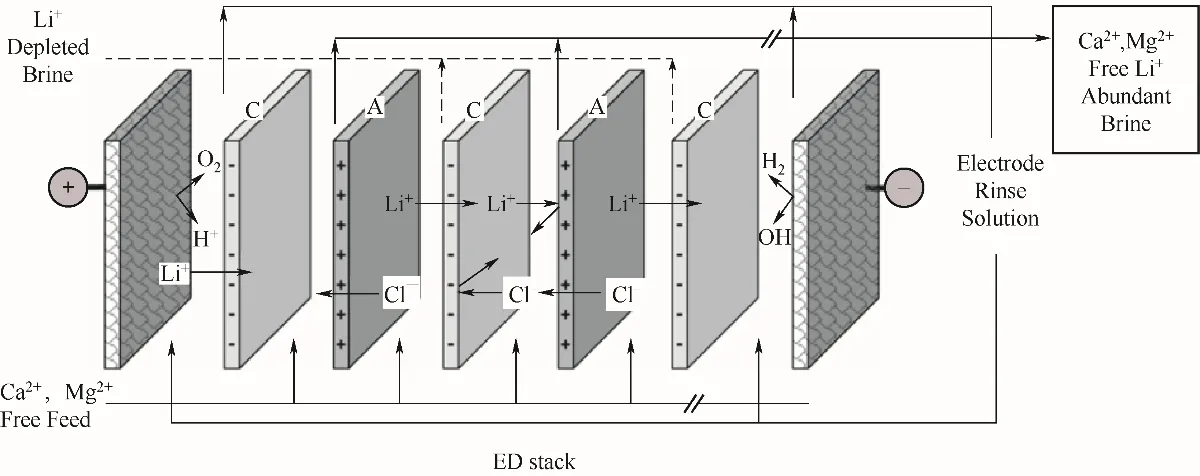
圖13 電滲析過程示意[73]Fig.13 Schematic diagram of the electrodialysis process[73]
Yu等[74]采用電滲析技術對不同進料特性的鹵水進行處理,評價了Selemion CSO膜的選擇性與親和性。在鹽水系統中發現一種特殊的轉移現象,證明該方法對各種鹵水中回收鋰具有適用性。天然鹵水實驗也表明,該技術能有效實現鋰回收。
選擇性電滲析(S-ED)是一種從較高Mg/Li比鹵水中分離回收鋰的新興技術。Yu等[75]采用單級SED工藝,研究了電流、溫度和鹵水總溶解固體(TDS)濃度對鋰回收率、選擇性遷移率、電流效率和能耗的影響。發現低于極限電流密度的大電流既能提高鋰回收率,又能提高分離性能。高溫下鋰、鎂離子分離度明顯降低,鋰回收率變化不大。采用四級分步調節電流的S-ED工藝,分離后Mg/Li質量比由9.85降至0.57時,鋰回收率達90%。
Zhao等[76]報道了一種液膜電滲析法,可以選擇性地從鹵水中回收鋰。液膜由離子液體和兩層固體陽離子交換膜組成,離子液體作為Li+的載體,對Li+具有較高選擇性和長期穩定性,兩層固體陽離子交換膜夾在載體上。利用電場輔助的夾心液膜可實現Li+識別和選擇性電遷移。在電流密度為4.375 A/m2的條件下,電滲析12 h后,Mg/Li比由初始鹽水的50∶1降至接收液的0.5∶1,電流效率65%和比能耗16 Wh/g均比典型電滲析工藝低。
Zhao等[77]進一步提出了一種結合液膜萃取和電滲析的夾心式液膜電滲析系統(圖14)。夾心液膜由兩層陽離子交換膜和一層負載鋰的有機液膜組成。選擇TBP-TABLO4-體系作為液膜,能夠將鋰離子與其他陽離子(Na+、K+、Mg2+、Ca2+)有效分離。該系統適應性強、能耗低,具有很好的應用前景。
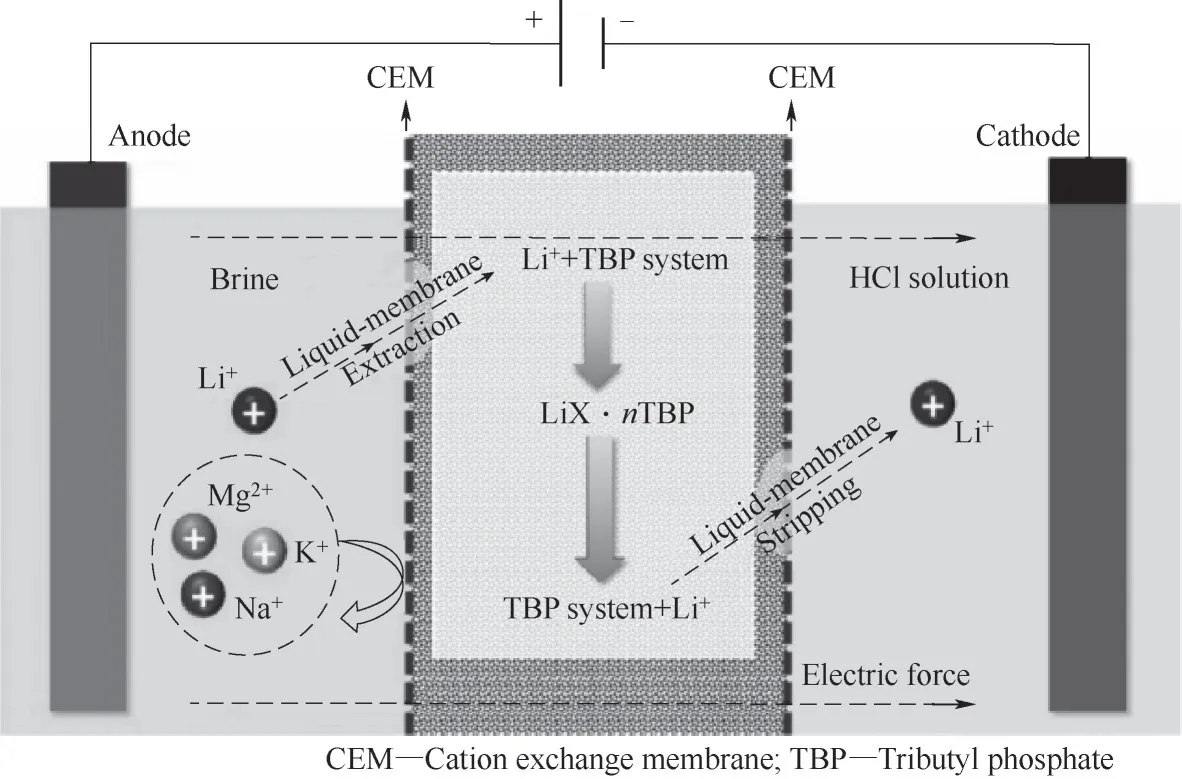
圖14 液膜萃取和電滲析夾心式液膜電滲析系統回收Li+[77]Fig.14 Liquid membrane extraction and electrodialysis sandwich liquid membrane electrodialysis recovery Li+system[77]
電滲析工藝能耗低(40~270 Wh/mol),可處理較高Mg/Li比和高Mg2+濃度的鹽湖鹵水。該工藝可在不同濃度的鹽湖鹵水中實現鎂/鋰分離。雖然單價選擇性離子交換膜的電滲析能有效去除二價離子,但從含有共存單價離子(Na+、K+等)的鹵水中有效回收鋰仍具有挑戰性。
4.3 雙極膜電滲析
雙極膜電滲析技術(bipolar membrane electrodialysis,BMED)是在普通電滲析系統中引入雙極膜,以促進水在電場下解離成H+和OH-的過程,在直流電場驅動下,OH–可以與從含鋰原料液遷移進入堿室的Li+結合形成氫氧化鋰,因此該方法在由含鋰溶液制備氫氧化鋰方面具有應用前景。目前研究側重于由鹽湖鹵水制備氫氧化鋰的工藝。針對我國鹽湖高鎂鋰比特點,可將雙極膜電滲析與其他鎂鋰分離方法相結合,制備較高純度的氫氧化鋰產品。該方法與傳統鹽湖氫氧化鋰制備技術相比,具有低能耗、綠色環保、產品品質高等特點,在提升鹽湖鋰鹽產品品質、豐富鋰產品種類方面具有一定優勢。
Xu等[73]利用電滲析對低鎂鋰比鹵水進行富集濃縮,再由沉淀法得到碳酸鋰,去除二價陽離子雜質,最后利用雙極膜電滲析制備氫氧化鋰。Shen等[78]采用離子選擇性電滲析與雙極膜耦合,利用選擇性離子交換膜分離二價雜質離子,一定程度上解決了二價陽離子對離子交換膜的結垢污染,但制備的氫氧化鋰液濃度低于0.2 mol/L,需進行高倍濃縮后才能進行氫氧化鋰結晶。從高Mg/Li比鹽湖鹵水制備氫氧化鋰面臨著分離鋰/鎂、富集鋰、生產高純度氫氧化鋰的挑戰。Wang等[79]采用雙極膜電滲析與NF、RO、CED相結合的一體化膜工藝,從Mg/Li比大于30的鹽湖鹵水中制取LiOH(圖15)。結果表明,該工藝制備的堿液鋰濃度大于1.0 mol/L,電流效率為36.05%,能耗為6.20 kWh/kg,是從鹽湖鹵水中制備氫氧化鋰很有發展前景的工藝。

圖15 BMED與NF、RO、CED一體化膜從鹽湖鹵水中制取LiOH[79]Fig.15 Schematic diagramof the production of LiOH fromsalt lake brine with integrated membranes of BMED,NF,ROand CED[79]
雖然膜分離技術是一種可行的工藝,但其發展受到高運行成本的限制。因此,膜技術的未來發展應包括提高分離效率、改善材料穩定性、膜污染控制和膜設計,而開發高選擇性膜材料至關重要。
5 電化學法
電化學方法利用鋰離子電池中的Li插層/脫層原理,工作電極作為鋰捕獲材料先從鹽水中捕獲Li+,再將其釋放到溶液中回收。在電化學作用下可避免脫鋰過程用酸洗脫材料,從而減少了材料溶損,增強了循環性能,是一種低能耗、高效率的提鋰技術[80-81]。在電化學方法中,要求鋰捕獲材料具有優良的選擇性、高的鋰容量和長期穩定性。
5.1 離子捕獲系統
Kanoh等[82-83]報道了利用電極材料從溶液捕獲鋰離子的電化學方法。該過程是基于可充電鋰離子電池的鋰插層/脫嵌機制,捕獲和釋放鋰離子的反應為:

Chen等[84]通過恒電位處理LiMn2O4合成了一種不需高溫和有機黏結劑的自支撐λ-MnO2電極,在電化學提鋰中表現出較高的鋰離子遷移能力,更大的容量和更好的循環性能,能耗約為4.14 Wh/mol Li+。Wang等[85]用石墨布改性LiNi0.6Co0.2Mn0.2O2制備核殼型微球材料(rGO/NCM),解決電極材料的腐蝕問題。電子在石墨布中傳輸,有效抑制了NCM晶格塌陷。由rGO/NCM和活性炭(AC)組成的脫鹽電池能提取93%的Li+,提取容量為13.84 mg/g,消耗能量1.4 Wh/mol。他們還研究了由LiNi0.038Mo0.012Mn1.95O4(LNMMO)和交流陽極組成的混合超級電容器從鹽水中提取鋰,提取容量為14.4 mg/g,97.2%的Li+被捕獲,消耗能量7.91 Wh/mol[86]。在鋰離子捕獲系統中,Li+在工作電極上被捕獲,陰離子在對電極上被捕獲。由于LiFePO4能夠在較寬pH范圍內的水溶液中保持穩定,具有良好的可逆性能,常被選為工作電極。當鋰被提取時,LiFePO4轉變到FePO4,通過施加反向電壓,AgCl被還原為Ag,實現鋰的提取回收。反應如下:
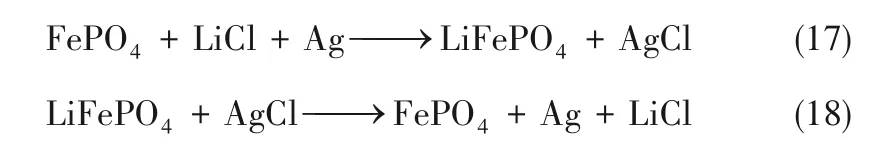
La Mantia等[87]報道了一種從鹽水中回收鋰的電池(圖16),該電池由捕獲Li+的陽離子電極(LiFePO4)和捕獲Cl-的陰離子電極(Ag)組成。回收每千克鋰消耗電能144 Wh,可將富鈉的鹽水(Li∶Na=1∶100)轉化為富鋰溶液(Li∶Na=5∶1)。

圖16 采用LiFePO4-Ag體系進行電化學鋰回收[87]Fig.16 LiFePO4-Agsystem for electrochemical lithium recovery process[87]
Zhao等[88]將磷酸鐵鋰電極材料充電機理與漿料電解工藝相結合,回收廢棄磷酸鐵鋰材料。在陰離子膜漿料電解過程無須添加化學試劑,可分離Li和FePO4。鋰浸出率達98%,而96%以上的鐵以FePO4/C被回收。
5.2 搖椅電池系統
搖椅電池是在電池充電和放電期間,鋰離子分別從電池的正、負電極插入和脫出,鋰離子的傳輸類似于搖椅運動。Li+從陰極室的鹽水中被選擇性捕獲;同時,Li+被釋放到陽極室。鋰離子在正極和負極中具有良好的可逆性。常利用LiFePO4/FePO4電極選擇性地從鹽湖鹵水中提取鋰,鹽水中Li+被FePO4選擇性捕獲。同時,Li+從LiFePO4里脫出并富集,反應式為:
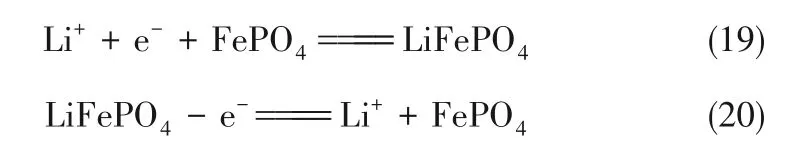
通過調節電位將插入負極中的Li+釋放到電解質中,陰離子交換膜阻止鋰離子從陽極室進入陰極室,從而達到鋰離子在陽極室中富集。循環結束后,交換正、負極后再運行,可以從鹽水中連續提取鋰。
Zhao等[89]依據搖椅電池原理從鹵水中提取鋰,構建了LiFePO4|NaCl溶液|陰離子交換膜|鹵水|FePO4電池體系(圖17)。在處理原鹵時,鹵水中Mg/Li比從初始的134.4降至陽極電解液中的1.2,鋰回收率83%。在處理老鹵時,Mg/Li比由初始的48.4降至0.5,陽極電解液中的鋰濃度上升為初始的6倍,從0.51 g/L上升到3.2 g/L,提鋰容量約為5 mg/g。使用電化學方法提取鋰的過程簡單、連續性好,回收過程不用添加其他化學試劑,幾乎不引入雜質,產品的純度和回收率高。但電極上的化學反應需避免副反應,對電解液組成要求較高,系統需要進一步優化,目前尚未規模化生產。
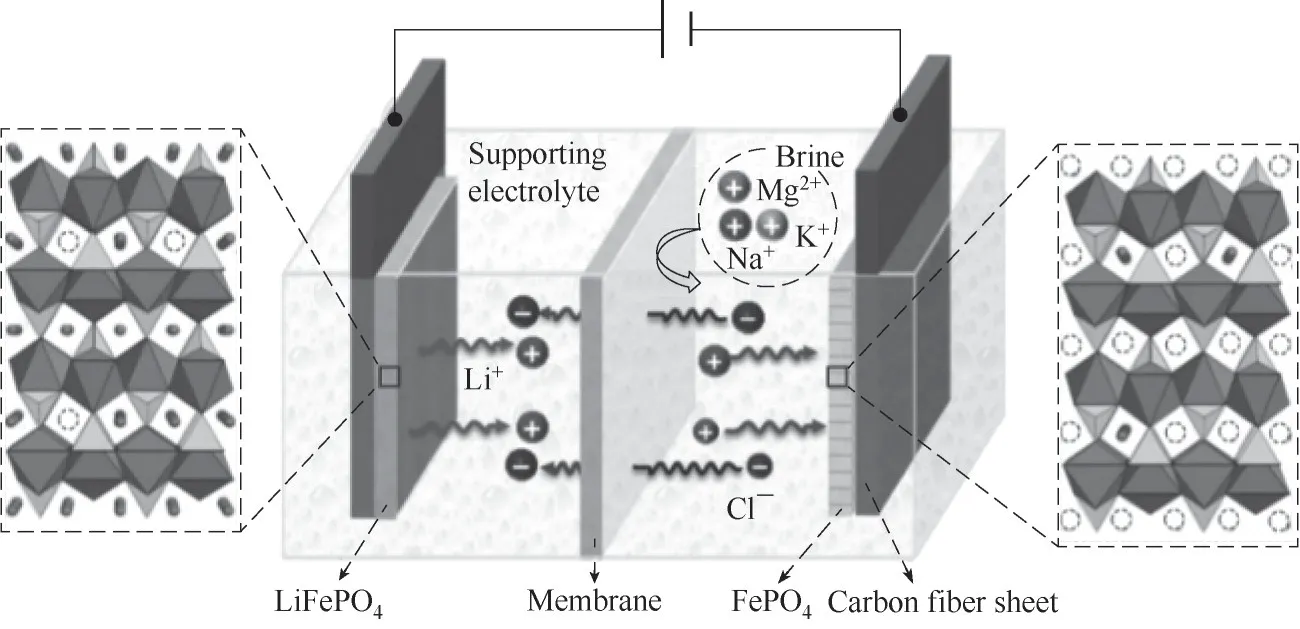
圖17 利用LiFePO4/FePO4結構電化學提取鋰[90]Fig.17 Structure of the LiFePO4/FePO4 for electrochemical lithiumextraction[90]
6 總結與展望
從高鎂鋰比鹽湖鹵水中提取鋰的主要方法有萃取、吸附、新興的反應/分離耦合技術以及以壓力驅動的膜分離法,而以電驅動的膜分離法及電化學法只適用于低鎂鋰比鹽湖鹵水鋰提取,對于高鎂鋰比鹽湖提鋰則需要與其他分離方法相結合。由于各鹽湖鋰資源稟賦特點存在差異,現階段以萃取、吸附、膜分離為主的鹽湖提鋰技術已分別在青海柴達木地區各鹽湖鋰鹽產區得到產業化應用,已在青海建成總產能近10萬噸/年的碳酸鋰生產線。本文對現有提鋰技術研究進展進行了系統綜述,并結合相關技術的產業化實施效果,對現有鋰提取技術方法的特點以及存在問題進行總結,見表2。
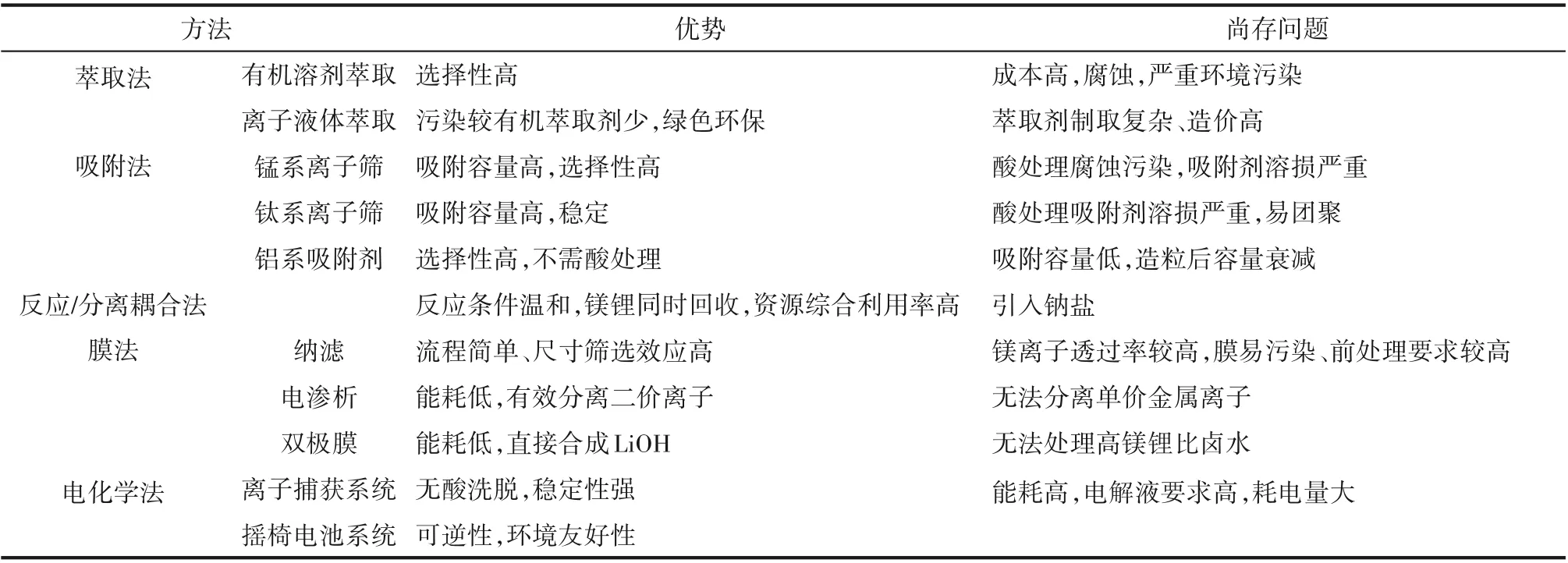
表2 現有鹽湖鹵水鋰提取技術比較Table 2 Summary of existing lithium extraction techniques
近年來,隨著我國青海、西藏鹽湖鹵水鋰提取技術的不斷發展,萃取法、吸附法等較為傳統的提取方法已經得到一定應用,而以納濾法、電滲析法為代表的膜法和反應/分離耦合技術也在逐漸興起,并受到廣泛關注。依據各分離技術的優勢和特點,耦合利用不同提鋰技術是未來青海地區鹽湖提鋰的一個重要發展方向,比如膜法與吸附法等其他技術組合,可進一步提升鎂鋰分離效果和鋰濃度,高鎂鋰比鹽湖鹵水可利用反應/分離耦合技術進行鎂鋰分離,之后再利用普通電滲析及雙極膜電滲析直接加工鋰鹽產品,而無須傳統的蒸發濃縮和化學法沉鋰技術,有利于提升鋰資源利用率、降低能耗。
基于目前我國青海鹽湖提鋰技術進展及產業化發展狀況,針對青海地區高鎂鋰比鹽湖鋰資源的開發利用,尚需解決如下問題:
(1)鹽湖鋰資源開發利用的總收率偏低;
(2)提鋰后資源的綜合利用程度低;
(3)鎂鋰分離技術有待優化提高,開發高純的氯化鋰、氫氧化鋰、金屬產品,延長鋰產品產業鏈,實現鋰資源高值化、多元化利用;
(4)鹽湖高效提鋰技術的工程化和產業化研究待加強。
由于地理環境、氣候條件以及光照條件等因素影響,西藏地區鹽湖鹵水蒸發速度低、老鹵產出周期長,因此,青海地區鹽湖提鋰技術未必適用于西藏鹽湖鋰資源的開發。針對西藏地區鹽湖鋰資源開發,應結合西藏地區的環保要求,發展低能耗、無污染、綠色環保的提鋰技術,因此,吸附法和膜分離技術在西藏鹽湖提鋰中具有突出的應用前景。總之,我國鹽湖主要位于青藏高原生態環境脆弱區,未來的鹽湖提鋰技術應充分利用當地的太陽能、風能、冷能和地熱等天然資源優勢,發展高效鹽田工藝,減少外來化學試劑添加。在開發鋰資源的同時,促進鹽湖資源的綜合利用,積極開發提鋰后高鎂或高鈉鹵水及沉鋰母液等高鹽廢水綜合治理及鋰資源再利用技術,促進鹽湖提鋰技術向高效、集約、綠色環保的方向發展。
