羥基積雪草苷對大鼠創傷性顱腦損傷后神經功能障礙的改善作用及機制研究
2021-07-23 06:34:30賈顏鋒
創傷外科雜志 2021年7期
關鍵詞:劑量
賈顏鋒,陳 偉
1.西安交通大學第一附屬醫院神經外科,西安 710061;2.西安交通大學附屬西安市人民醫院神經外科,西安 710004
在創傷性顱腦損傷(traumatic brain injury,TBI)后繼發的復雜病理生理進程中,受損腦組織產生的興奮性氨基酸過量聚集,可通過作用于神經元細胞膜N-甲基-D-天門冬氨酸 (N-methyl-D-aspartic acid,NMDA) 受體,引起興奮性神經毒損傷和細胞凋亡,是造成TBI后神經功能障礙的重要因素[1-2]。研究表明,細胞表面的NMDA受體2B亞基 (NMDA receptor 2B,NR2B) 磷酸化水平與表達量越高,其對興奮性氨基酸的毒損傷易感性越強[3-4]。因此,抑制TBI后NR2B與磷酸化NR2B (Phosphorylated NR2B,pNR2B) 表達水平,可能是阻止NMDA受體介導的興奮性神經毒損傷以及連鎖效應的有效途徑。
羥基積雪草苷(madecassoside,MC)是提取自傘形科植物積雪草的三萜類化合物,是積雪草的主要藥理活性成分,具有抗氧化應激、抗炎、促進創面愈合等多種功效[5-6]。新近研究發現,MC對中樞神經系統退行性病變和缺血性疾病具有神經保護作用[7]。然而,MC對顱腦損傷的實驗研究未見報道。本文通過建立大鼠TBI模型,觀察MC對顱腦損傷后神經功能障礙的改善作用,并從興奮性毒損傷效應引起神經元凋亡的角度初步探討MC的可能作用機制,為其應用于TBI的腦保護治療提供新的研究依據。
材料與方法
1 主要設備與試劑
控制性皮層損傷打擊裝置(美國Hatteras 公司);免疫印跡電泳裝置(美國Bio-Rad公司);NR2B抗體和pNR2B抗體(武漢博士德生物工程有限公司);B細胞淋巴瘤-2(B cell lymphoma,Bcl-2)基因抗體、半胱天冬酶-3(Caspase-3)抗體和Bcl-2相關X蛋白(Bcl-2 associated X,Bax) 抗體(美國CST公司);β-actin抗體(美國Thermo Fisher公司);Western-blotting檢測試劑盒和BCA蛋白定量試劑盒(北京碧云天生物科技有限公司);羥基積雪草苷 (南京道斯夫生物科技有限公司)。
2 實驗分組與給藥方法
SPF級雄性SD大鼠64只(200~220g/只),由西安交通大學醫學部實驗動物中心提供。采用隨機數字表法將大鼠分為四組:假創傷組、創傷組、創傷后低劑量MC治療組(25mg/kg)、創傷后高劑量MC治療組(75mg/kg)。MC溶解于0.9%氯化鈉注射液中,低劑量MC組與高劑量MC組大鼠在傷后1h時尾靜脈注射給藥干預,此后每日在相同時點尾靜脈給藥1次,連續28d。本研究大鼠飼養和實驗過程均嚴格遵守實驗動物倫理規定,獲西安交通大學醫學部動物實驗倫理委員會批準(XJTU-M202001295a)。
3 建立大鼠TBI模型
以2%戊巴比妥鈉溶液腹腔注射麻醉大鼠(60mg/kg),沿中線縱行切開頭頂皮膚,剝離骨膜,于人字縫尖與前囟的中點右偏4mm處,牙科鉆開一直徑為3mm的顱骨骨窗,顯露硬腦膜。連接控制性皮層損傷裝置,將大鼠固定于打擊臺上,調整打擊棒位置,使其尖端接觸至硬腦膜表面,實施皮層打擊致傷,打擊速度為4.0m/s、致傷深度2.0mm、接觸時間為120ms,完成打擊后予以縫合頭皮。假損傷組大鼠僅縱形切開頭皮后縫合,不予以實施打擊。
4 蛋白免疫印跡檢測(Western-blotting)
大鼠傷后28d處死,采用Western-blotting檢測各組大鼠皮層損傷區NR2B、 pNR2B、Bcl-2、Caspase-3和Bax的表達水平。以2%戊巴比妥鈉溶液腹腔注射麻醉大鼠(60mg/kg),快速斷頭取腦,冰上分離獲得皮層損傷區腦組織。冰浴條件下裂解、勻漿、提取總蛋白。采用蛋白質定量法(BCA法)蛋白定量后,上樣進行SDS聚丙烯酰胺凝膠電泳、轉膜。5%脫脂奶粉溶液封閉1h后,TBST洗滌3次,每次8min。分別加入NR2B一抗(1∶1000)、pNR2B一抗(1∶800)、Bcl-2一抗(1∶2000)、Caspase-3一抗(1∶1000)、Bax一抗(1∶2000)和β-actin一抗(1∶1500),4℃條件下反應過夜,TBST洗滌3次,每次8min。再加入IgG二抗(1∶2000),室溫條件下反應1h,TBST洗滌3次,每次8min。加入顯影液進行發光,凝膠成像系統分析,測定各目標條帶與內參β-actin條帶的灰度,得出相對灰度比值。
5 大鼠神經功能評分
傷后3、7、14、21和28d,采用改良神經功能缺損嚴重程度評分量表(modified neurological severity score,mNSS) 分別對各組大鼠神經功能進行評估。根據該量表評分標準與步驟,依次在不同時點對各組大鼠進行運動實驗、感覺實驗、平衡木實驗和反射實驗[8]。綜合各項實驗得分,分數越高代表神經功能缺損越嚴重,1~6分為輕度功能缺損,7~12分為中度功能缺損,13~18分為重度功能缺損。
6 統計學分析
結 果
1 大鼠神經功能評分比較
傷后3、7、14、21和28d,創傷組、低劑量MC組與高劑量MC組大鼠在各時點的mNSS評分均高于假創傷組(P<0.05)。傷后3d,創傷組、低劑量MC組與高劑量MC組的評分差異無統計學意義(P>0.05)。傷后7、14、21和28d,低劑量MC組與高劑量MC組的評分均顯著低于創傷組(P<0.05),并且高劑量MC組評分顯著低于低劑量MC組,組間差異有統計學意義(P<0.05)。此外,在傷后7、14、21和28d,低劑量MC組與高劑量MC組的評分隨著時間延長逐漸降低,兩組內各時點間的差異也有統計學意義(P<0.05)。見表1。

表1 各組大鼠傷后各時點的mNSS評分比較分)
2 大鼠皮層損傷區NR2B與pNR2B表達水平
傷后28d,創傷組大鼠皮層損傷區的NR2B與pNR2B表達量較假創傷組顯著增加。與創傷組相比,低劑量MC組、高劑量MC組兩個給藥組的NR2B與pNR2B表達水平明顯降低。并且高劑量MC組的NR2B與pNR2B表達水平均顯著低于低劑量MC組,組間差異有統計學意義(P<0.05)。見圖1、表2。
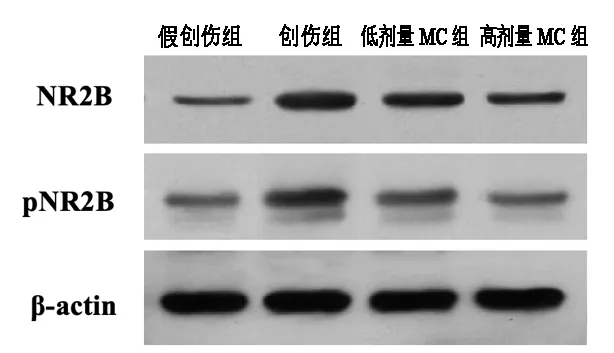
圖1 各組大鼠皮層區NR2B與pNR2B表達檢測結果
表2 各組大鼠NR2B與pNR2B表達量比較
3 大鼠皮層損傷區Caspase-3、Bax與Bcl-2表達水平
與假創傷組相比,創傷組大鼠皮層損傷區域的Caspase-3、Bax與Bcl-2的表達量均增加。與創傷組相比,低劑量MC組與高劑量MC組的Caspase-3與Bax表達水平減少,Bcl-2表達水平明顯增加。兩個給藥組相比,高劑量MC組Caspase-3與Bax的表達水平顯著低于低劑量MC組,Bcl-2的表達水平高于低劑量MC組,組間差異存在統計學意義(P<0.05)。見圖2與表3。

圖2 各組大鼠皮層區Caspase-3、Bax與Bcl-2表達檢測結果
表3 各組Caspase-3、Bax與Bcl-2表達量比較
討 論
隨著現代社會和交通工具的發展,TBI作為常見的急性創傷,發生率逐漸升高。TBI以高致殘率和致死率居各類創傷之首,已成為嚴重威脅公共健康的衛生問題。雖然救治技術的進步使TBI病死率顯著下降,但對幸存患者的神經功能障礙救治難以令人滿意,尤其是對TBI后多因素參與的繼發性腦損傷缺乏有效干預措施[9]。因此,深入研究TBI后繼發性腦損傷的致傷因素與機制,探討合理可行的治療策略,對改善神經功能缺損和促進患者康復具有重要意義。本研究利用控制性皮層損傷法制備TBI模型,初步發現MC對TBI具有神經保護作用。
MC作為傳統中藥積雪草的主要藥理成分,既往研究證實其對中樞神經系統多種退行性疾病具有神經保護作用[5]。MC可顯著提高側索硬化小鼠的脊髓前角神經元數量,減少神經元變性,有效改善側索硬化小鼠的神經功能缺陷[7]。MC通過抑制1甲基-4-苯基-1,2,3,6-四氫吡啶(MPTP)對多巴胺能神經元的毒性損傷作用,減少神經元的凋亡,增加紋狀體多巴胺的含量,顯著改善帕金森大鼠的運動功能障礙[10-11]。MC還可抑制腦缺血再灌注損傷時小膠質細胞向促炎表型極化,減輕小膠質細胞介導的炎癥反應,減少促炎因子的釋放和自由基的產生[12-13]。大鼠脊髓損傷后,MC干預可顯著提高脊髓組織的超氧化物歧化酶活性,減輕損傷脊髓的水腫程度和神經元凋亡數量[6]。
在TBI后繼發性腦損傷的病理生理變化中,損傷區域腦組織興奮性氨基酸大量聚集,結合至位于神經元細胞膜的N-甲基-D-天冬氨酸(NMDA)受體,而引起興奮性神經毒損傷效應,導致神經元細胞凋亡、壞死,進而加重繼發性腦損害[2-4]。構成NMDA受體的多個亞單位中,主要調節亞單位NR2包括NR2A、NR2B、NR2C和NR2D四個亞基。其中,NR2B亞基作為調控NMDA受體功能與活性的主要分子,其磷酸化是NMDA受體介導興奮性神經毒損傷過程中的關鍵環節,直接決定了神經元細胞對興奮性氨基酸毒損傷效應的易感性[14-15]。研究表明,抑制NR2B的表達及其磷酸化水平,可有效減輕興奮性氨基酸堆積引起的細胞外鈣離子內流超載,以及最終導致的興奮性毒損傷效應[4,16]。
本研究利用控制性皮層損傷法建立TBI大鼠模型,首先通過mNSS神經功能缺損評分,證實了TBI引起的運動、感覺、反射等神經功能障礙。給予MC治療發現受傷大鼠的神經功能障礙明顯改善,而且高劑量MC治療組的效果顯著優于低劑量MC治療組。皮層損傷區NR2B與pNR2B的檢測結果發現,TBI后NR2B表達水平及其磷酸化水平顯著增加,表明傷后皮層區NR2B被活化,引起興奮性神經毒性損傷,可能是導致TBI后神經功能障礙和加重繼發性腦損傷的重要因素。給予MC干預治療,可顯著抑制NR2B的表達及其磷酸化,改善大鼠的神經功能障礙。同樣,筆者發現,高劑量MC對NR2B介導的興奮性神經毒性損傷抑制效果顯著優于低劑量。
神經元凋亡是受興奮性毒損傷后的主要死亡原因之一,抑制TBI后神經元細胞凋亡發生、發展,可有效減輕繼發性神經損害和促進功能恢復[17-18]。在目前已知的眾多調節細胞凋亡分子中,Bcl家族的促進凋亡基因Bax和抑制凋亡基因Bcl-2是一對極為重要的調控分子。Bax與Bcl-2在凋亡進程中的對立功能,使二者的表達水平直接決定了細胞凋亡閾值[19]。在凋亡信號刺激下,Bax構象發生改變,并轉位至線粒體破壞膜通透性,引起大量促凋亡因子釋放,激活Caspase-3,進而啟動細胞凋亡和加速凋亡進程[20]。Bcl-2作為抑制細胞凋亡基因,通過與Bax形成異源二聚體,可抑制其轉位以及活化Caspase-3的級聯反應,有效阻止細胞凋亡程序啟動[21]。Bcl-2還可通過調節內質網中鈣離子流向,穩定胞內鈣離子濃度,組織鈣離子超載進一步加重,阻斷興奮性毒損傷誘發的細胞凋亡進程[19-21]。
如何減輕興奮性毒損傷引起的神經元凋亡,是近年來TBI的腦保護策略研究熱點。本研究在明確TBI后皮層損傷區NR2B介導的興奮性毒損傷基礎上,進一步對該區域凋亡相關分子進行檢測,結果提示傷后促凋亡分子Caspase-3和Bax表達明顯增加,抗凋亡分子Bcl-2表達顯著下降,表明NR2B活化引起的興奮性毒效應可誘導神經元細胞凋亡。傷后予以大劑量和小劑量MC干預治療,均能夠有效抑制NR2B活化和神經毒性反應,下調促凋亡分子表達和提高抗凋亡分子的表達,逆轉TBI后興奮性毒損傷引起的神經元凋亡,并且高劑量組的效果優于低劑量組。據此,筆者推測,MC對顱腦損傷的神經保護作用,可能與減輕NR2B介導的興奮性神經毒損傷及其引起的神經元凋亡有關。
綜上所述,本研究通過建立大鼠TBI模型和給予MC干預治療,發現TBI后MC可有效抑制興奮性神經毒損傷的重要誘導分子NR2B活化,減輕皮層區神經元凋亡,改善神經功能障礙,初步證實了MC對TBI具有神經保護作用,為其向臨床轉化應用提供了更加充分的實驗依據。但TBI后MC的最佳治療劑量、給藥時間窗和具體作用途徑等仍有待進一步研究。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
今日農業(2022年4期)2022-11-16 19:42:02
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
藥學與臨床研究(2015年4期)2015-06-05 11:35:54
衛生職業教育(2014年24期)2014-05-20 09:05:38
同位素(2014年2期)2014-04-16 04:57:20
中國合理用藥探索(2014年11期)2014-03-11 20:30:20
