生物質基含氧化合物在過渡金屬碳化物上加氫脫氧研究進展
2021-07-24 08:58:46方輝煌吳歷潔陳偉坤袁友珠
化工學報 2021年7期
關鍵詞:催化劑
方輝煌,吳歷潔,陳偉坤,袁友珠
(廈門大學化學化工學院,固體表面物理化學國家重點實驗室,醇醚酯化工清潔生產國家工程實驗室,能源材料化學協同創新中心,福建廈門361005)
引 言
生物質作為唯一可再生的碳資源,利用生物質制取高品質的液體燃料和化學品,對緩解資源短缺、減少環境污染具有重要意義,已成為一個重要發展方向[1-2]。例如,對于由木質素和纖維素以及半纖維素組成的木質纖維素,是生物質一大種類,人們已發展多種技術途徑實現木質纖維素的資源化利用,包括水解、熱解、液化和木質素優先轉化等[3–6]。這其中,木質纖維素快速熱解生產生物油是最經濟有效的初步解聚途徑[7-9],其主要利用在少量或隔絕空氣的情況下,經過超高加熱速率(1000~10000℃/s),將木質纖維素經常壓瞬間氣化并快速凝結成液體,從而得到以含氧化合物為主的初級生物油(圖1)。如圖1所示,木質纖維素經過快速熱解制得由眾多含氧化合物組成的生物油;但生物油存在含氧基團多、熱穩定性差、熱值低、黏度大和腐蝕性強等問題,難以直接利用,需要進一步在催化劑的作用下,通過氫解、加氫、脫氧和HDO 等C—O 鍵活化和轉化,從而制得高品質生物基燃料、化學品和延伸的生物基材料[10-11]。
涉及生物質基C—O 鍵活化和轉化的催化劑有大量文獻報道,主要有貴金屬和非貴金屬材料。常見的貴金屬催化劑有Pt、Pd、Ru、Rh、Re 和Ir 等[12-16]。然而,貴金屬價格昂貴,儲量少,限制了貴金屬催化劑的實際應用。非貴金屬催化劑主要是Ni基、Co基和Cu 基催化劑[17-23]。例如,Cu 基催化劑廣泛用于糠醛氣相加氫制糠醇、油脂加氫制脂肪醇和甲醇合成等領域[18-19];Ni 基催化劑常用于木質素酚類化合物等諸多加氫/氫解反應中[20-23]。過渡金屬碳化物是一類間隙型化合物,由碳原子在金屬晶格中的插入調變了母體金屬的電子能帶結構,從而具有類貴金屬的催化性質,是一類廉價高效的催化材料[24]。過渡金屬碳化物具有優異的C—O 鍵活化能力,被廣泛用于加氫、脫氧、氫解等反應體系中[25-28]。近幾年,關于金屬碳化物應用于生物質基含氧化合物HDO反應中的研究報道不斷增多。
本文在簡要分析了生物質熱解油中代表性含氧化合物的結構特性和反應路徑的基礎上,綜述了應用于這些含氧化合物HDO 反應過渡金屬碳化物的制備、結構和構效關聯,并對該研究領域進行了總結與展望。雖然另一類主要來源于植物油脂,以硬脂酸和油酸為代表的生物質基含氧化合物,在實際應用過程中往往也需進行HDO 處理,但限于篇幅,此處不作贅述。
1 生物質熱解油組成與結構特性

圖1 木質纖維素催化轉化制備燃料、生物材料和高附加值化學品Fig.1 Production of bio-fuels and value-added chemicals by transformation lignocellulose

表1 典型生物油的組成成分[29-31]Table 1 Major components in typical pyrolysis bio-oil[29-31]
表1列舉了典型生物質熱解油的具體組成與含量,其組分十分復雜,包括醇類、醛類、酮類、酯類和芳香醚酚等400 多種含氧化合物[29-31]。這些生物質基含氧化合物中存在大量的酚羥基、醇羥基、羰基和醚鍵等不同形式的C—O 鍵,直接限制其進一步的工業應用,需要進一步HDO 提質[32-34]。在生物質熱解油中,最具代表的是愈創木酚和苯甲醚等芳香酚類醚類、糠醛等呋喃類以及小分子乙酸和丙酮等高含氧化合物,它們常被當作經HDO 途徑實現生物質高值利用研究的模型底物,以研發催化劑的結構與性能關系,為制備附加值更高的燃料和化學品奠定基礎[35]。
表2示出了生物質熱解油含氧化合物中典型的C—O 化學鍵及其解離能[36]。不同的C—O 鍵解離能差異較大,其平均解離能高達358 kJ·mol-1,這使得C—O鍵活化與轉化的反應路徑增多且不易調控,對催化劑作用機理的理解和高選擇性催化活性中心結構的構筑帶來困難。為升值轉化這些含氧化合物,其重要途徑是通過加氫、氫解和HDO 等催化反應得以實現。加氫反應是使不飽和C O鍵和C C鍵在氫氣作用下發生加成反應,不同催化劑的活性位與C O 鍵和C C 鍵的相互作用不盡相同,進而實現對C O 鍵和C C 鍵選擇性加氫;氫解反應是在氫氣作用下,將C—O 鍵斷裂并重新形成C—H 鍵的過程[37],化合物中不同基團連接的C—O鍵具有不同的解離能,同樣要求催化劑對不同的C—O 鍵類型具選擇性;而HDO 則是包含C O/C C鍵加氫和C—O鍵氫解等過程,研發兼具高活性和高選擇性的HDO催化劑是該領域的一個關鍵科學問題。

表2 典型的生物質基C—O 鍵及其解離能[36]Table 2 Typical biomass based C—O bonds and their dissociation energies[36]
2 過渡金屬碳化物的結構、性質和制備
過渡金屬碳化物在許多涉氫反應中表現出與Pt、Pd和Ir等貴金屬相似的加氫脫氧性能,廣泛用于包含生物質熱解油在內的各類含氧化合物的HDO反應中。過渡金屬碳化物種類繁多,結構多樣,其獨特的催化性質來源于滲碳引起的結構變化,這些結構差異直接影響其性質和催化性能。為此,本小節簡述過渡金屬的結構性質,以及近些年來發展的制備方法和結構特性。
2.1 金屬碳化物的結構與性質
過渡金屬碳化物是一類由碳原子填充在母體金屬晶格的四面體空穴中形成的間隙型合金。其結構主要有面心立方堆積(fcc)、六方密堆積(hcp)和六方堆積(hex)三種結構(圖2)[38]。這些晶體的幾何結構遵循H?gg 規則,由半徑比(radius ratio)rc/rm決定,其中rc和rm分別為碳原子和過渡金屬原子的半徑。當rc/rm小于0.59 時,則金屬碳化物形成以fcc、hcp和hex為主的常見結構[39-40]。例如,第四族和第五族的過渡金屬形成的主要以fcc結構為主的晶體結構,有TiC、ZrC、NbC、VC 和TaC 等。而第六族形成的碳化物晶體結構就更為復雜,W2C 主要以hcp結構存在,而WC 則是為hex結構。對于碳化鉬而言,β-Mo2C,η-MoC 和γ-MoC 具有相似的hcp結構,而α-MoC1-x則具有fcc結構。
過渡金屬碳化物不僅具備良好導電性,而且具有較高的熔點和硬度,因而往往展現特別的催化產物選擇性和催化性能穩定性[41-42]。1973 年,Levy等[43]報道了碳化鎢具有類鉑催化特性,使其作為一種潛在的替代貴金屬催化材料引起了人們的極大興趣。碳原子在母體金屬中的插入使得母體金屬晶格發生膨脹,金屬原子之間間距拉大,導致金屬d帶寬化,進而造成態密度發生變化。這些費米能級處態密度的重新分布將對不同平面取向和吸附物質產生明顯影響。由于碳原子s-p 軌道與過渡金屬原子的s-p-d 軌道的相互作用,使得過渡金屬碳化物中存在由共價鍵、離子鍵和金屬鍵混合的三種不同相互作用[39-40],這些相互作用所帶來的電子結構性質變化,使得碳化物具有獨特的催化性質。

圖2 過渡金屬碳化物的晶體結構和第Ⅳ~Ⅷ族的金屬碳化物種類Fig.2 Crystal structure of transition metal carbides and the normal metal carbides in Group Ⅳ—Ⅷ
2.2 金屬碳化物的制備
過渡金屬碳化物的制備方法也將直接影響碳化物的結構與特性[44]。其中,程序升溫碳化法是制備過渡金屬碳化物最為常見的方法[45]。該方法主要是采用過渡金屬氧化物為前體,以氣體碳氫化合物為碳源,在程序升溫的條件下,碳氫化合物在高溫下解離為碳原子滲入到母體金屬晶格中(圖3)。在常用的氣體碳源中,以甲烷/氫氣、乙烷/氫氣、丁烷/氫氣等混合氣為主[46-47]。Claridge 等[48]研究了程序升溫碳化法對不同金屬碳化物的影響,發現該方法在制備碳化物中具有一定的普適性。Xiao 等[49]通過研究碳化物制備方法的影響,發現碳化物納米粒子的晶相和結構很大程度上取決于反應條件。對該方法而言,升溫速率、碳化溫度和氣體流速都直接影響到金屬碳化物的形貌、結構和催化活性。在程序升溫碳化法中,氣體碳源和固體氧化物間發生氣固界面的快速滲碳反應,對于碳化物的晶相結構往往不易控制;高溫碳化燒結引起低比表面積,同時還容易生成包裹碳層,減少碳化物活性中心暴露,直接影響了碳化物在催化等方面的性能。
隨后發展起來的碳熱還原法表現出了制備高分散和高比表面積碳化物的優越性。該方法采用具有高比表面積的碳載體材料作為碳源,如活性炭(AC)、碳 納 米 管(CNT)和 碳 納 米 纖 維(CNF)等[41-42,50-54]。這些碳載體利用浸漬法負載金屬前驅鹽,再在高溫下進行還原碳化。由于固-固界面的滲碳反應有效降低了碳載體中碳原子的擴散,減少了碳化物表面石墨碳層的生成,有利于活性位點的暴露。同時,由于碳載體具有較大的比表面積和孔道結構,有利于金屬前驅鹽的分散,從而制得具有高分散的碳載體負載型碳化物材料。基于該方法,Kim 等[55]合成了還原石墨烯負載型具有高分散的Mo2C、Fe3C、WC 和W2C 催化劑,并發現這些碳化物的粒徑小于它們自身的金屬氧化物前體,說明了還原石墨烯碳載體在碳熱還原中有利于納米粒子的重構,生成均勻分散的碳化物顆粒。Liang 等[56]利用碳熱還原法在700℃的氫氣中合成了活性炭負載的均勻的β-Mo2C/AC 催化劑,其粒子尺寸在10 nm 左右。Han 等[57]采用CNT 作為載體制備出具有hcp結構的20%Mo2C/CNTs,其粒徑在10.5 nm 左右。相似地,Jongerius 等[58-59]則是采用CNF 合成了Mo2C/CNF和W2C/CNF 催化劑并應用于愈創木酚HDO 反應。這些CNT 和CNF 作為載體具有比表面積大、允許活性組分均勻分散、化學穩定性高、耐強酸堿環境以及特殊的纖維結構等優勢,有利于催化活性的進一步提升。

圖3 過渡金屬碳化物的制備方法Fig.3 Preparation methods of transition metal carbides
近些年來,除了常見的碳載體用于碳熱還原法制備碳化物外,一些含碳的金屬有機無機化合物也被用于金屬碳化物的制備中。其中,最為典型的為金屬有機框架材料(MOFs)[60-63]。在制備過程中,這些有機配體在高溫下熱解,生成碳原子并滲入到周圍的金屬的晶格中。由于MOFs 具有較大的比表面積和豐富的孔道結構,往往可以制備介孔碳化物。Wu 等[61]采用銅基MOFs(HKUST-1)作為前體,并將Mo 雜多酸限閾在MOFs 中。經過高溫滲碳處理和Cu 刻蝕處理,得到具有豐富介孔的碳負載型MoCx催化劑。相似地,Shi等[62]在合成納米MoC 的過程中使用MOFs {Mo3(BTC)2},得到3 nm 左右的碳化物材料。Wan等[63]通過選擇不同的MOFs,分別以ZIF-67和ZIF-8 作為前體,合成了具有一維和二維結構的Mo2C。除了MOFs 以外,Yu 等[64]利用羧基聚苯乙烯微球作為模板在引入Mo 前驅鹽的條件下合成三維有序介孔碳化鉬。Fang等[65]則是采用金屬有機聚合物的方式,在偏鎢酸銨的存在下,利用甲醛與間苯二酚聚合的方式形成樹脂前體,在經過碳化后可以得到碳球負載的均勻碳化鎢顆粒。綜上可見,研究者針對不同的應用場合采用不同的制備策略,進而達到最優效果。可以預料,針對包括生物質在內各種碳資源優化利用的需求,人們將不斷研究與開發更為先進的碳化物制備方法。
3 生物質熱解油含氧化合物加氫脫氧反應
3.1 芳香醚酚化合物的加氫脫氧
在生物質熱解生物油中,芳香醚類和酚類化合物是最常見的、含量最高的含氧化合物,如何對這些化合物高效加氫脫氧是生物油提質的關鍵。作為生物油中最典型的化合物,愈創木酚含有3 種不同的C—O 鍵(Ar—OCH3,ArO—CH3和Ar—OH),常被研究者作為HDO 反應的研究對象[4,11,66-67]。圖4 示出了以愈創木酚和苯甲醚為例的反應路徑圖。整個過程以HDO反應為核心,通過催化劑設計與反應條件的調變,從而控制反應過程中的O/C 和H/C 比,以獲得預期產物。愈創木酚HDO 的反應路徑主要有兩種,一種是優先進行C—O 鍵的氫解反應,包括芳香基C—O 鍵的直接氫解生成苯酚、脂肪基C—O鍵氫解生成鄰苯二酚,從而進一步氫解生成苯酚和苯;這些化合物可能進一步加氫得到環己醇、環己酮、環己烯和環己烷等產物。而另一種反應路徑則是優先進行苯環的加氫反應,進而發生C—O 鍵氫解反應。例如,Mo2C/CNF 和W2C/CNF 在愈創木酚HDO 的反應中,優先氫解芳香基C—O 鍵,高選擇性得到苯酚[58]。又如,NiMo/Al2O3-SiO2催化劑在反應溫度較低時,生成以脂肪基C—O 鍵斷裂為主的鄰苯二酚產物;當升高反應溫度時,則得到以芳香基C—O 鍵斷裂為主的苯酚產物[68]。Zhao 等[69]采用Pd/C 與磷酸雙功能催化劑,在水相條件下催化愈創木基化合物優先進行芳香環加氫,進而發生C—O 鍵氫解,生成環烷烴。這些均表明了反應路徑取決于催化劑性質和反應條件的控制。
表3 列舉了過渡金屬碳化物在芳香醚和酚類HDO 反應中的研究結果[53,58,70-83]。Bitter 團隊[58]報道了過渡金屬碳化物如Mo2C/CNF 和W2C/CNF 用于愈創木酚和苯甲醚的HDO反應,發現碳化物具有較好的C—O 鍵解離能力。其中,Mo2C/CNF 顯示出更高的愈創木酚氫解活性,在350℃和5.5 MPa H2條件下,其苯酚收率達到67%。在愈創木酚氫解中,不同的碳化鉬催化劑,如Mo2C@C、MoC1-x/CNF、α-MoC1-x/AC、Mo2C core/shell 等,在一定的反應條件下均表現出了優異的C—O 鍵氫解能力,得到以苯酚和烷基酚為主的產物,其收率在49%~91.8%之間[70-73]。Fang 等[74]通過固體碳源碳熱還原法合成晶相可控的碳化鎢催化劑,并用于生物質基醚酚的化合物的氫解中。活性結果顯示,特定W2C/WC 比例組成的金屬碳化鎢催化劑表現出優異的選擇性斷裂芳香基C—O 鍵的能力,而不進行芳香環的加氫,可將愈創木酚、苯甲醚、二甲氧基苯酚等轉化為氧含量更低的芳香化合物。Guo等[75-76]報道W2C/AC 能夠對木質素模型化合物進行選擇性C—O 鍵氫解,通過β-O-4 解離得到收率較高的木質素生物油。除了愈創木酚外,過渡金屬碳化物在苯酚、烷基酚和苯甲醚等含氧化合物中亦表現出獨特的芳香基C—O鍵的活化能力,得到以苯和烷基苯等為主化學品[77-82]。例如,Boullosa-Eiras等[79]系統研究了二氧化鈦負載的金屬碳化鉬、氮化鉬、磷化鉬和氧化鉬在苯酚氫解的性能。結果表明15%(質量)Mo2C/TiO2在350℃和2.5 MPa 的氫氣壓力下能高效氫解苯酚得到苯,其選擇性高達90%。由上所述,過渡金屬碳化物在這些芳香類含氧化合物中通過HDO 和氫解,得到更低含氧量芳香化合物,是利用生物油制備高附加值燃料和化學品的有效途徑。
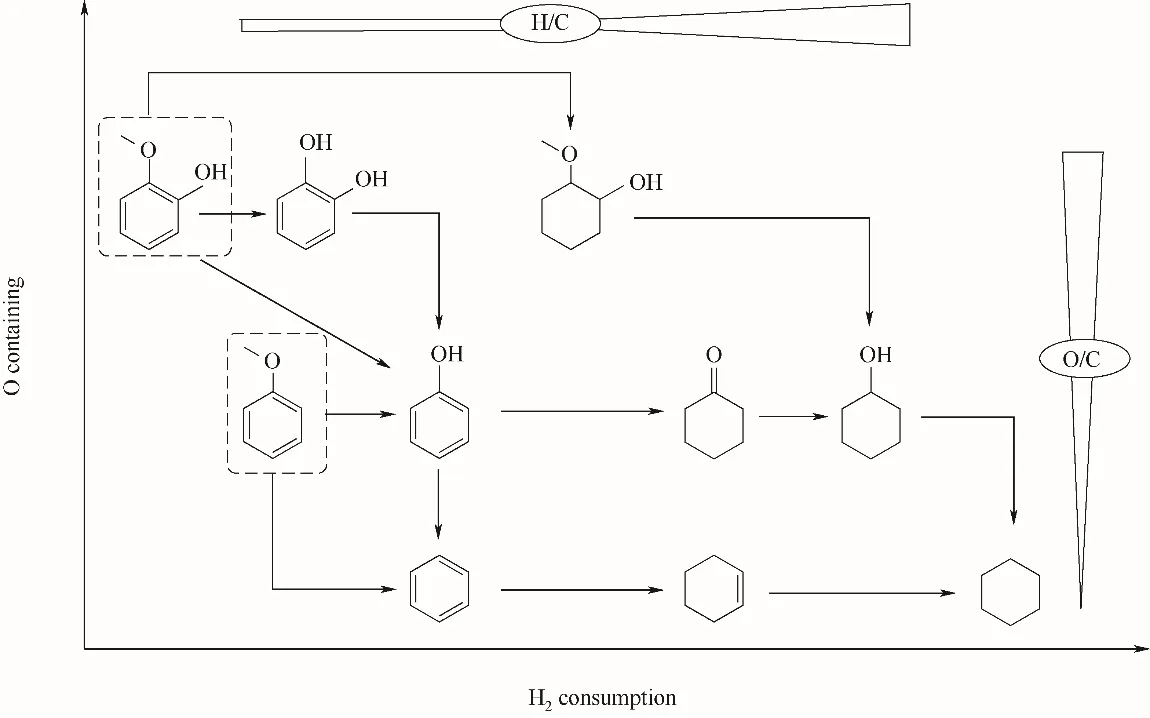
圖4 愈創木酚和苯甲醚加氫脫氧的反應路徑Fig.4 Reaction pathway of guaiacol and anisole hydrodeoxygenation
3.2 其他含氧小分子的加氫脫氧
除芳香醚類和酚類化合物外,生物油中還含有大量的小分子酸、醛、酮、醇和酯類化合物。其中,比較有代表性的有乙酸和乙醛等C2、丙酮和丙醛等C3、呋喃醛和環戊酮等C4和C5含氧化合物。這些分子雖然具有較短的碳鏈,但卻擁有C O 和C—O 鍵等豐富的含氧官能團。如何實現選擇性HDO 反應亦是對這些高含氧化合物加氫提質的關鍵。同時,由于這些小分子具有簡單的化學結構,也常被研究者作為模型化合物展開研究。
過渡金屬碳化物對小分子醛酮酸的HDO 反應可展現出獨特的催化性能[84-93]。表4 列舉了若干過渡金屬碳化物用于生物質基酸、醛、酮和醇等小分子含氧化合物HDO 的反應結果。Schaidle 等[84]通過程序升溫反應考察了Mo2C 在乙酸HDO 中的催化性能,得到以乙醛和乙烯為主的產物。他們發現表面部分氧化的碳化鉬同時含有活化氫的金屬位點和含有羥基的布朗斯特酸性位,協同催化C—O 鍵氫解。進而,他們制備了SBA-15負載的Mo2C催化劑,納米顆粒尺寸在1.9 nm 左右[85]。Bhan 團隊[86]研究了Mo2C 在丙酮HDO 中的催化性能。丙酮首先在碳化鉬的金屬位點加氫生成異丙醇,并在布朗斯特酸性位上發生脫水反應生成丙烯;丙烯可能進一步發生加氫反應生成丙烷。Ren等[87]合成介孔Mo2C用于丙醛的加氫脫氧,得到以丙烯為主的不飽和碳氫化合物。此外,過渡金屬碳化物還被用于以糠醛為模型化合物的呋喃類含氧化合物的HDO 中,得到以2-甲基呋喃為主的反應產物[89–91]。從這些文獻結果可見,過渡金屬碳化物往往表現出獨特C O 鍵加氫和C—O 鍵氫解能力,而對C—C 鍵不發生氫解反應,從而可獲得較高附加值的HDO產物。
3.3 真實生物質熱解油的加氫脫氧
從上文可知,過渡金屬碳化物應用于生物質熱解生物油模型化合物HDO反應已取得相當進展,可將各類模型化合物高效地轉化為燃料和化學品。在此基礎上,研究者們將這些碳化物的催化應用從單一模型底物拓展到混合底物,研究其在真實生物質熱解油HDO 中的構效關聯。例如,直接以初級生物油為反應原料,考察金屬碳化物等催化劑在反應中的轉化效率、產物選擇性和穩定性等。Chen 等[94]將苯甲醚、愈創木酚、甲酚和1,2-甲氧基苯酚等典型的生物質基含氧化合物混合作為底物,在Mo2C/CNF催化劑的作用下進行HDO 反應,在280℃下混合底物的轉化率為94%,其中苯、甲苯和環己烷的選擇性分別為66%、27%和5%。Li 等[95]采用磷化鎳將苯酚、苯甲醚和愈創木酚的混合物催化得到以苯和環己烷為主的產物。Remón 等[96]則直接用Mo2C/CNF將熱解的初級生物油用于HDO中,并同時考察了反應溫度、氫氣壓力和反應時間的影響,在不同反應條件下可以得到收率在17%~72%的提質生物油,其中混合酚、環酮化合物、羧酸化合物、酯和其他芳香化合物分別占56%~78%、7%~30%、2%~8%、0~9%和0~20%。這些研究表明,過渡金屬碳化物具有潛在的工業應用價值。盡管如此,相比于模型化合物,真實生物油中組分復雜,含氧量更高,這使得碳化物保持優異的C—O 鍵氫解能力具有一定難度,需要發展新的催化策略和方案。

表3 芳香醚和酚類化合物的加氫脫氧Table 3 Hydrodeoxygenation of aromatic ethers and phenols

表4 生物質基含氧小分子的加氫脫氧Table 4 Hydrodeoxygenation of simple oxygenates
Fang等[97]采用耦合雙功能催化劑的策略用于真實生物油HDO 反應。如圖5 所示,在芳香醚酚化合物的HDO 過程中,碳化鎢具有較好的芳香基C—O鍵氫解能力,但其活化氫氣的能力較弱,難以催化芳香環飽和反應。Ni 基催化劑活化氫氣能力較強,能同時對C—O 鍵氫解和芳香加氫,但催化選擇性較差。因此,把Ni 基催化劑與碳化鎢進行耦合,得到雙功能催化劑。結果顯示,該雙功能催化劑對愈創木酚HDO 具有高活性。當兩個催化劑均勻復合時,其活性達到最高,反應產物主要為飽和烷烴;當改變兩個催化劑的耦合方式和組分時,可實現產物的選擇性切換,分別得到環己醇或環己烷主產物。進一步的實驗結果表明,該雙功能催化劑的高轉化性能主要緣由兩個催化劑的協同效應得以實現,碳化鎢主要負責酚類分子中的C—O 鍵氫解,而Ni 則負責芳香環的加氫,并拉動反應的進行。該雙功能催化劑在各類生物質酚類化合物的HDO 反應中,同樣表現出優異的催化活性。基于這些結果,將其應用到真實的生物油反應中,得到碳收率高達45%的液態烷烴。Routray 等[98]采用Ru/C-Pt/ZrP 二步催化體系對熱解生物油HDO,得到30%的總碳收率;Duan 等[99]采用Pd/m-MoO3-P2O5/SiO2雙功能催化劑用于水溶性生物油的HDO 中,得到約為46.3%碳收率的液態烷烴。相比于這些貴金屬催化體系的結果,這種Ni基催化劑與碳化鎢耦合的催化體系具有一定的優勢和應用前景。
3.4 碳化物催化劑作用機理
為深入理解金屬碳化物在含氧化合物中HDO的作用機理,研究者對碳化物的表面表征和構效關聯展開深入的研究。Li 等[100]研究了金屬氮化物和碳化物的H2–TPD 行為。研究發現,這些催化劑的表面室溫下表現出對氫物種較強的吸附行為,從而生成不可逆的吸附氫。這些吸附氫物種在300~500℃的溫度區間才發生脫附行為。通過一系列的表征,研究認為氫吸附的活性位可能是配位不飽和的相中心。圖6提出可能的氫吸附和活化機理。首先,H2分子擴散至碳化物表面,然后這些H2分子在配位不飽和的金屬中心M 上活化解離,生成M—H和C—H 物種。這些配位不飽和的金屬中心是碳化物滲碳過程中由于碳空穴缺陷形成的。表面生成的M—H 鍵的鍵能較低,容易發生斷裂從而從催化劑表面遷移到吸附的反應底物中發生反應。此時,帶負電荷的氫物種(C—H)也會轉移至此表面或者晶格間隙中。這主要是由于C—H 鍵的鍵能遠大于M—H 鍵,在此表面的C 元素占支配地位,從而使得表面和此表面的氫物種達到一個平衡狀態。當表面金屬M 的原子濃度較高時,H2也可能在M—M 上面發生均裂。在整個反應過程中,氫氣分子的活化就像是一個儲氫容器一樣,在催化劑活性位點上不斷地進行氫活化、轉移和補充。
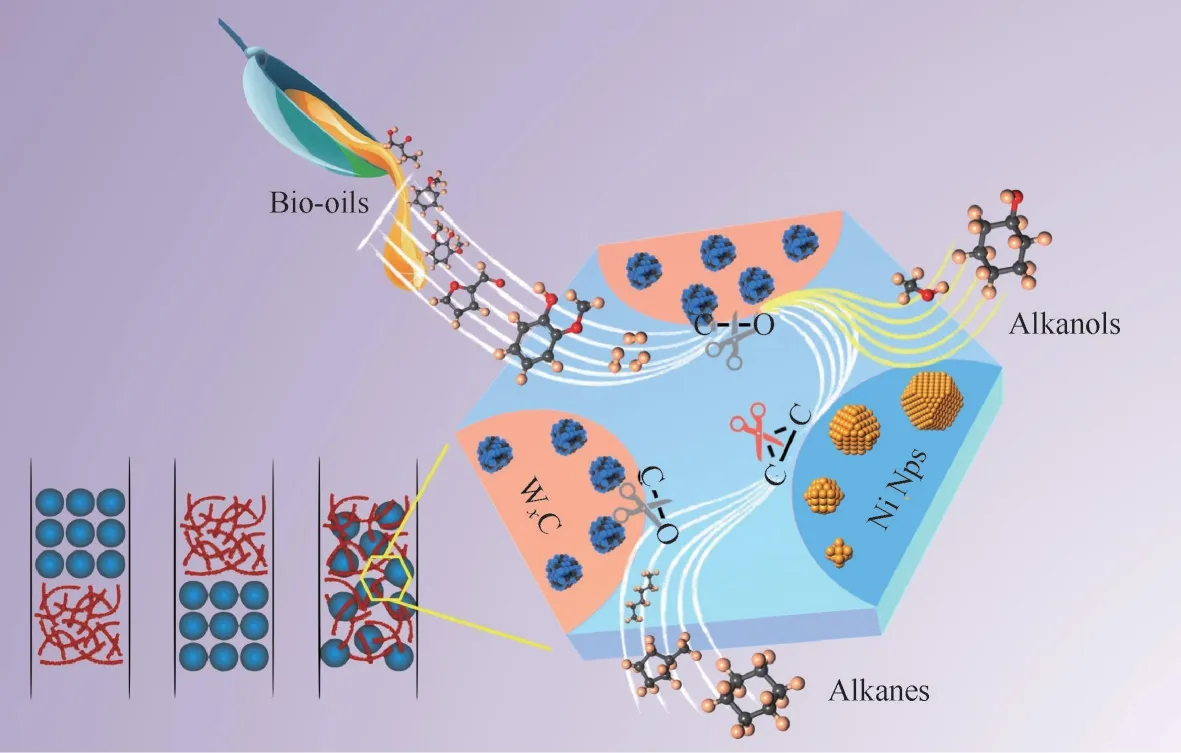
圖5 耦合雙功能催化劑用于生物油加氫脫氧Fig.5 Hydrodeoxygenation of biomass-derived bio-oil over integrated dual catalysts

圖6 氫氣在過渡金屬碳化物表面的活化解離機理Fig.6 Activation and dissociation mechanism of hydrogen on the surface of transition metal carbides

圖7 愈創木酚在過渡金屬碳化物表面的氫解機理Fig.7 Hydrogenolysis mechanism of guaiacol on the surface of transition metal carbides
金屬碳化物除了對H2分子的活化吸附以外,對反應底物分子的活化也起到關鍵的作用。以愈創木酚為例,圖7 示出了愈創木酚在金屬碳化物表面的催化機理。金屬碳化物表面先將H2分子解離為活潑氫物種。由于碳化物表面的金屬M 具有較強的親氧性,愈創木酚的甲氧基以線式吸附的形式吸附在表面位點。活潑的氫物種遷移到愈創木酚中,通過斷裂Caryl—OCH3鍵,生產吸附的苯酚中間物種[101]。生成的甲醇分子脫附或部分進一步發生氫解生成甲烷和水。而苯酚中間物種通過吸附形式的變換,在催化劑表面發生脫附生成苯酚或者進一步發生氫解從而生成苯和水。后續的C—O 鍵氫解取決于催化劑的表面活性和反應條件。上述反應機理表明,在金屬碳化物中,表面活性位點的暴露對C—O 鍵氫解的催化活性起著至關重要的作用。Fang 等[74]研究了碳化鎢不同相變化對愈創木酚活化能的改變以及對產物選擇性的影響,發現特定的滲碳使碳化鎢對C—O 鍵氫解有較高的選擇性,得到以苯酚為主的產物。為進一步研究活性位點的構效關聯,對碳化鎢進行表面重構,并量化了表面C/W 原子比與反應速率的關系,發現當C/W 比為7.2 時,其C—O 鍵氫解速率最高。這主要是由于隨著碳化鎢表面的重構,表面暴露出更多的碳空穴,即不飽和配位的金屬活性中心,有利于氫氣分子和含氧底物分子的活化。Liu 等[53]通過合成具有大量碳空位的W2C 納米棒,并用于苯唑呋喃的脫氧反應中,發現碳空穴活性位點的暴露有利于選擇性C—O 鍵的氫解,得到較低含氧量的高附加值化學品。
圖8 以乙酸、乙醛和糠醛等為例闡述了生物油的其他小分子含氧化合物在碳化物上的HDO 機理。這些分子一般含有C—O 或C O 鍵。在碳化物表面,這些含氧基團以線式或面式吸附于活性金屬位點;另一方面,活化的氫物種進而去進攻而發生加成或解離反應。生成的不飽和C C 可能進一步加氫生成飽和烷烴。例如,Ren等[87-88]報道了丙醇和丙醛在WC 表面的脫氧機理。丙醇經羥基解離線式吸附在WC 上,而丙醛則采用C O 雙鍵面式吸附,進而發生氫解反應,得到丙烯產物。他們還考察了Mo2C 的活性,發現除丙烯外還有丙烷的生成,這是因為Mo2C 比WC 具有更強的加氫能力。Shi 等[102]通過DFT計算糠醛在Mo2C的不同吸附形式,其中最穩定的是C O 雙鍵順式吸附在催化表面。Xiong等[103]通過TPD 等表征研究糠醛脫氧的反應機理,發現其經加氫反應得到糠醛中間體,進而氫解得到2-甲基呋喃。

圖8 含氧小分子在過渡金屬碳化物表面的氫解機理Fig.8 Hydrogenolysis mechanism of simple oxygenates on the surface of transition metal carbides
4 過渡金屬碳化物催化劑穩定性
盡管金屬碳化物在生物質熱解油含氧化合物的HDO中表現出優異的催化性能,其催化性能可通過調控碳化物結構得以進一步優化,但碳化物催化劑在這些HDO 反應中的穩定性是備受關注另一科學問題[104]。對失活機理的深入認識有助于開發出更高效穩定的碳化物基催化材料。目前,對于金屬碳化物催化劑的失活,普遍的因素包括:納米粒子團聚燒結、碳化物表面氧化、活性物種溶出和積炭等[50]。如圖9 所示,對于納米粒子的團聚燒結,是小顆粒碳化物催化劑中普遍存在的問題。雖然小顆粒碳化物具有大量暴露的催化活性位點,但是較高的表面能使得小顆粒傾向于通過遷移和Ostwald 熟化形成大顆粒,尤其在催化環境下,反應的溫度、氣氛和催化劑本身的分散情況等都直接影響著粒子的團聚長大。另一個因素是碳化物表面的氧化。碳化物在HDO中,其表面暴露在氫氣和含氧底物分子中,催化過程涉及氧化還原反應,當表面的金屬位點與含氧物種中的氧原子結合過強,就容易形成氧元素在催化劑表面的累積,形成金屬氧化物。Lee等[91]通過原位實驗在HDO 中觀察到了氧化碳化物的生成。其他的報道也顯示了反應過程中可能生成金屬氧化物或氧化碳化物等物種[50,92,105]。Boullosa-Eiras 等[79]在進行苯酚HDO 反應的研究中也發現,隨著反應的進行,Mo2C/TiO2發生失活現象,并伴隨著環己烯選擇性的升高,這是由于C—O 鍵氫解比加氫反應更容易失活引起的。活性物種溶出一般發生在液相反應體系中,在特定的反應條件下,活性金屬位點可能發生脫落并溶解于溶液中,伴隨著活性中心數量的下降,造成催化劑失活。而對于積炭行為,一般發生在催化裂解和氫解反應。盡管有研究指出,碳化物基催化劑在失活之后能夠通過一定的還原處理和碳化處理可以使得催化劑再生,恢復一定的催化活性,但是如何設計出高效穩定的碳化物催化劑依然是該領域研究的重點,有待深入研究和認識。
5 結 語
綜上所述,生物質熱解油中含氧化合物的HDO是制備生物燃料和高值化學品的重要催化反應,這些含氧化合物中C—O 鍵的反應路徑較為復雜,為高選擇性地獲得目標產物,減少副反應發生,必然要求對催化劑活性位點進行精確構筑。對于生物質熱解油代表性含氧化合物的HDO反應,反應底物主要可分為芳香基含氧醚酚、小分子酸醛酮和呋喃類化合物,反應產物主要取決于催化劑種類和催化劑活性位點結構。總體而言,在碳鏈含氧化合物的HDO 反應中,由于碳化鉬具有較強活化氫分子的能力,在碳化鉬上的HDO 容易得到飽和直鏈烷烴,而碳化鎢對氫分子活化能力適中,在碳化鎢上的HDO可獲得以不飽和產物為主的烯烴等產物;在芳香環含氧酚類醚類化合物的HDO反應中,過渡金屬碳化物具有優異的芳烴C—O 鍵氫解能力,可得到苯酚、苯和烷基苯等高附加值化學品。
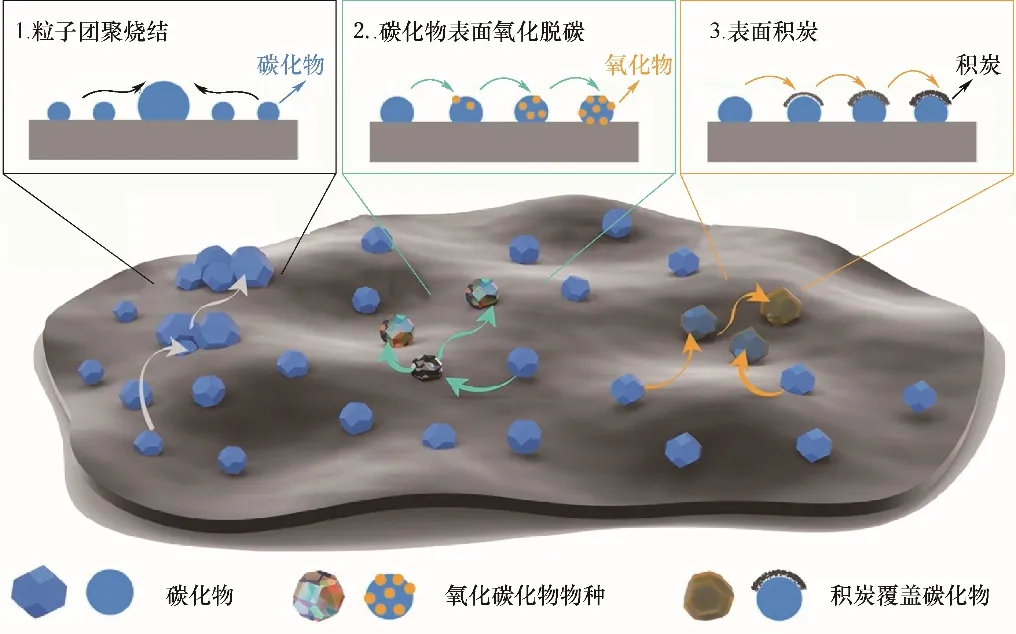
圖9 過渡金屬碳化物在催化反應中失活的可能機制Fig.9 Possible mechanism of deactivation of transition metal carbides in catalytic reaction
過渡金屬碳化物是由碳原子通過擴散進入到母體金屬的晶格間隙中,因而制備方法對其結構和表面有重要影響。程序升溫還原法雖然是制備碳化物的成熟方法,但得到的碳化物往往是體相催化劑,活性表面低且表面容易因為碳沉積而形成碳層包裹。碳熱還原法能夠通過固固界面反應降低碳原子擴散速率和表面碳累積,暴露更多的金屬活性位點,進一步提高活性;常用的碳源有活性炭、碳納米管和碳納米纖維等,改進后的方法還引入了一些含碳的有機-無機雜化物進行制備金屬碳化物,如MOFs 等。盡管如此,如何制備高效過渡金屬碳化物催化劑依然是該研究領域的關鍵,仍有不少問題需要解決。如圖10 所示,比較突出的科學問題有:(1)碳化物催化劑有別于金屬催化劑,其活性中心往往是由金屬原子和碳原子共同構筑,受到結構本體和表面中金屬/碳比的影響,在活性中心與C—O鍵活化中的性能強化作用機理和物理化學本質不夠清晰;(2)對于金屬負載型碳化物催化劑而言,許多文獻報道同金屬負載于不同晶型的碳化物中,在涉氫C—O 鍵活化反應體系表現出差異巨大的催化活性,理解這類體系中金屬與碳化物載體的強相互作用特性與共性,特別是各自晶體結構的關系、負載金屬與碳化物的匹配性等,值得進一步探究;(3)碳化物基催化劑的穩定性依然是一個亟待解決的問題,包括粒子團聚、表面氧化、積炭等。所有這些均是需要重點關注的問題。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
