表面改性未焙燒TS-1固載金催化丙烯氫氧環氧化反應性能研究
2021-07-24 08:58:46張志華杜威段學志周興貴
化工學報 2021年7期
張志華,杜威,段學志,周興貴
(華東理工大學化學工程聯合國家重點實驗室,上海200237)
引 言
環氧丙烷(PO)是目前僅次于聚丙烯的第二大丙烯衍生物,主要用于生產聚氨酯、表面活性劑、丙二醇等化工產品[1-2]。目前工業上PO 的生產方法主要有氯醇法和Halcon 共氧化法[2-4]。前者存在環境污染嚴重、廢水和廢渣排放量大和設備腐蝕等問題,后者存在工藝流程長、投資大和經濟性受聯產產品市場影響較大等問題[1-2,5]。過氧化氫液氧化法雖然具有較高的丙烯轉化率和PO 選擇性,但是該方法以昂貴的雙氧水為原料,且雙氧水需要現場生產,生產工藝較復雜,再加上后續的產品分離需要用到大量的蒸汽,導致該工藝的生產成本居高不下[5]。Haruta等[6]發現采用沉積-沉淀法(DP法)制備的Au/TiO2催化劑能夠在氫氣和氧氣氣氛下催化丙烯直接氣相環氧化生成PO,他們認為該反應過程的進行需要金和鈦的協同效應,氫氣和氧氣在納米金顆粒(<5 nm)上反應生成的HOOH物種溢流到Ti位點上形成Ti—OOH 中間體,然后丙烯再和Ti—OOH 物種發生環氧化反應生成PO[7-9]。與其他工藝相比,丙烯氫氧環氧化制備PO 工藝具有環境友好、操作簡單、產物易分離和選擇性高等優點,日益受到研究人員的廣泛關注,成為當今研究領域的熱點[1]。
目前,多種含鈦載體(如TS-1、Ti-MCM-41、Ti-SBA-15、Ti-MCM-48、Ti-TUD、Ti-MWW)固載的金催化劑被應用于丙烯氫氧環氧化反應中[10-16]。其中TS-1由于具有豐富的孤立四配位鈦物種,將納米金顆粒固載在TS-1 上制備的Au/TS-1 催化劑顯示出優異的催化活性[10-16]。然而,即使是TS-1 固載的納米金催化劑仍然存在穩定性差的問題,嚴重制約著丙烯氫氧環氧化工藝的工業化應用[17]。本課題組[18]研究發現反應中產生的焦炭堵塞TS-1 微孔孔道是導致Au/TS-1催化劑快速失活的主要原因。基于該失活機理,采用微孔孔道被模板劑堵塞的未焙燒鈦硅分子篩TS-1-B 作為載體,以氫氧化鈉為沉淀劑通過DP 法將金顆粒固載在TS-1-B 的外表面。采用該方法制備的Au/TS-1-B 催化劑在30 h 的考評時間內未發現明顯的失活現象,顯示了較好的穩定性[18]。值得指出的是,TS-1 分子篩在晶化過程中不可避免地會產生一些缺陷而形成Si—OH 等缺陷位硅物種[19],這會導致親水性的PO 在催化劑表面的羥基上吸附而發生開環、異構及聚合等反應生成導致催化劑失活的焦炭物種[20-22]。因此,若能對TS-1-B進行改性處理來降低表面缺陷位硅羥基的數量將有助于進一步提高催化劑的穩定性。
除含鈦載體外,催化劑制備方法也對Au-Ti 雙功能催化劑的性能具有重要影響。目前,DP法是制備Au-Ti 雙功能催化劑最常用的方法,該方法制備的Au-Ti 雙功能催化劑活性較高,但是金的負載效率通常較低(<3%)[17,23],并且制備過程煩瑣,大大限制了催化劑的工業放大。此外,該方法制備的催化劑不可避免地會殘留堿金屬離子[24],這可能會因堿金屬離子的殘留量難以控制而對Au-Ti協同效應的研究帶來干擾。Lu 等[25]發現以尿素為沉淀劑可以將大部分氯金酸固載到TS-1-B 分子篩上,金的負載效率大于90%,遠高于傳統的DP 法,且該方法制備的Au/TS-1-B 催化劑在丙烯氫氧環氧化反應中表現出優異的性能。因此,采用尿素為沉淀劑的DPU 法不但能夠獲得較高的金固載效率,還能夠排除堿金屬離子的干擾,更有利于研究Au-Ti 協同的本質。
采用有機堿溶液對TS-1 進行后處理改性來改善其催化性能已成為催化領域的一個研究熱點。研究發現采用四丙基氫氧化銨(TPAOH)溶液對TS-1 分子篩進行二次晶化改性時發生了溶解-再結晶過程,改性后的TS-1上產生了有利于分子擴散的晶內不規則介孔,并且改性處理過程中TS-1結構進一步完善,減少了硅羥基等缺陷位,提高了TS-1 表面疏水性,從而顯著提升了TS-1 分子篩的催化性能[26-28]。后續研究還發現采用四丁基氫氧化銨(TBAOH)、哌啶(PI)及四丙基溴化銨(TPABr)與乙胺(EA)的混合溶液對鈦硅分子篩進行二次晶化改性處理均有助于改善鈦硅分子篩的擴散性能并提高表面疏水性[26-27,29]。目前,采用有機堿溶液對未焙燒鈦硅分子篩TS-1-B 進行二次晶化處理后再固載納米金顆粒催化丙烯氫氧環氧化反應的研究尚未見報道。本文以尿素為沉淀劑采用DPU 法將納米金顆粒固載于TPAOH 溶液二次晶化改性的TS-1-B上,并以同樣方法將納米金顆粒固載在未改性TS-1-B 上作為參比催化劑,比較研究了這兩種催化劑在丙烯氫氧環氧化反應中的催化性能差異,并結合傅里葉變化紅外光譜(FT-IR)及高角度環形暗場-掃描投射電子顯微鏡(HAADF-STEM)等表征以建立催化劑結構和性能之間的關系。此外,本文還研究了二次晶化處理改性TS-1-B 固載金催化劑在丙烯氫氧環氧化反應中的動力學特性。
1 實驗材料和方法
1.1 材料
實驗用水為超純水,電阻為18.2 MΩ。吐溫-20,梯希愛(上海)化成工業發展有限公司。TPAOH,上海邁瑞爾化學技術有限公司提供,質量分數為25%。正硅酸四乙酯(TEOS),上海麥克林生化科技有限公司提供,純度> 99%。異丙醇(IPA),上海麥克林生化科技有限公司提供,ACS 光譜級。鈦酸四丁酯(TBOT),賽默飛世爾科技(中國)科技有限公司提供,純度>99%。氯金酸水合物,天津希恩思生化科技有限公司提供,純度(質量分數)為47.8% Au。尿素,上海泰坦科技股份有限公司,純度> 99%。氮氣(純度99.995%),氫氣(純度99.99%),氧氣(純度99.99%),丙烯(純度99.5%),液化空氣(昆山)氣體科技有限公司。
1.2 未焙燒鈦硅分子篩制備與改性處理
按照文獻[18]所述方法制備未焙燒鈦硅分子篩TS-1-B:首先將2.00 g 吐溫-20 滴加到28.60 g 超純水中,室溫磁力攪拌1 h 后逐滴滴加22.60 g TPAOH,滴加完TPAOH 后再磁力攪拌1 h,然后再逐滴滴加40.53 g TEOS,待加入TEOS 攪拌至澄清后再將0.62 g TBOT 與10.00 g 異丙醇混合液緩慢滴加到上述澄清液中,攪拌1 h 后將混合液在80℃下加熱除醇5 h,再將母液移入帶有聚四氟乙烯內襯的不銹鋼水熱釜,然后放入170℃的烘箱中晶化48 h。晶化結束后將水熱釜從烘箱中取出,待降至室溫后將漿液離心、洗滌后置于80℃恒溫烘箱中干燥6 h,最終制備出硅鈦比為100 的未焙燒鈦硅分子篩TS-1-B。
未焙燒鈦硅分子篩TS-1-B 的改性處理按如下方法進行[27]: 將TS-1-B 分子篩加入TPAOH 水溶液中,配比為1 SiO2∶0.04 TPAOH∶20 H2O,室溫下攪拌成均一漿液后倒入帶有聚四氟乙烯內襯的不銹鋼水熱釜中,然后將水熱釜置于恒溫烘箱(170℃)中晶化24 h。晶化結束后將水熱釜從烘箱中取出,然后在冷水中快速降至室溫,再將漿液離心、洗滌后置于80℃恒溫烘箱中干燥6 h,即得改性處理的未焙燒鈦硅分子篩TS-1-B-TPAOH。
1.3 催化劑制備
按照文獻[25,30]所述方法制備Au-Ti 雙功能催化劑:先將一定量氯金酸水溶液(0.96 g/L)與1.00 g TS-1-B-TPAOH 加入盛有40 ml 水的燒杯中,再加入0.09 g尿素,避光條件下連續磁力攪拌,并將該懸濁液從室溫升溫至90℃,繼續攪拌6 h 后將漿液置于50 ml 離心管中離心10 min,離心后用40 ml 水洗滌并再次離心,再將所得固體樣品置于真空干燥器中,在室溫下干燥12 h 后即得Au/TS-1-B-TPAOH催化劑。通過調節氯金酸的加入量可制備不同金負載量的催化劑。Au/TS-1-B催化劑的制備步驟與Au/TS-1-B-TPAOH 催化劑制備步驟相同。其中,0.09% Au/TS-1-B 及0.09% Au/TS-1-B-TPAOH 催化劑在制備時加入1000 μl 氯金酸溶液,0.08% Au/TS-1-B-TPAOH 催化劑在制備時加入850 μl 氯金酸溶液。
1.4 表征
鈦硅分子篩的物相分析在Rigaku D/Max2250 VB/PC 衍射儀(Rigaku公司,日本)上完成,測定條件為管電流100 mA,管電壓40 kV,入射光源為Cu 靶,Kα射線(λ=0.154056 nm),掃描速率12 (°)/min。在Perkin-Elmer Lambda 35型紫外-可見漫反射光譜儀(Plerkin-Elmer 公司,美國)上表征鈦硅分子篩的鈦物種,以BaSO4為參比。在Nicolet 6700 型傅里葉變換紅外光譜儀(Nicolet 公司,美國)上表征鈦硅分子篩的鈦物種及硅羥基,鈦硅分子篩樣品采用KBr 壓片。采用掃描電子顯微鏡(SEM)表征鈦硅分子篩的形貌與尺寸大小,樣品分析在JSM360LV(JOEL 公司,日本)上進行。在JEM 2100F型高分辨率透射電子顯微鏡(HR-TEM)(日本JOEL 公司)上分析鈦硅分子篩的形貌,加速電壓200 kV,儀器點分辨率0.23 nm,線分辨率為0.14 nm。在ASAP 2020 型氣體吸附測試儀(Micromeritics 公司,美國)上表征鈦硅分子篩的孔容、比表面積等織構性質。在液氮溫度(-196℃)下測定樣品的氮氣吸附-脫附等溫線,吸附前樣品在170℃下抽真空預處理。采用Brunauer-Emmett-Teller(BET)模型計算樣品的比表面積,樣品的總孔容由比壓為0.99 時所吸附的氮氣體積換算得到。采用等離子體-原子發射光譜測定催化劑上金的負載量。測試前將催化劑溶于王水與氫氟酸組成的混合溶液中,制備出含有一定濃度的金溶液后再進行分析。在IRIS 1000 型原子吸收光譜儀(Thermal Elemental 公司,美國)上進行金負載量的測定,分析波長175~1050 nm。采用高角度環形暗場-掃描投射電子顯微鏡(HADDF-STEM)分析納米金顆粒的粒徑分布及平均粒徑。HAADFSTEM 分析在Tecnai G2 F20 S-Twin(FEI 公司,荷蘭)上進行,最大加速電壓200 kV,放大倍數110 萬倍,點分辨率0.24 nm,晶格分辨率0.102 nm。
1.5 催化劑考評
采用常壓固定床實驗裝置(圖1)評價催化劑性能。該裝置的反應器為電加熱的連續流動常壓固定床反應器,該反應器由石英管、不銹鋼殼層、保溫層、加熱設備以及中空銅管這幾部分組成,其中最外層為不銹鋼殼層,保溫層位于不銹鋼殼層和中空銅管中間,內徑約為0.6 mm 的石英管放置于中空銅管中。催化劑裝填量為0.15 g(采用篩網孔徑0.15 mm 篩分),催化劑裝入石英管后通入H2/N2(36%(體積)H2,50 ml/min)混合氣體從室溫以1℃/min 的升溫速率升溫至200℃,然后再切換為原料氣(C3H6/H2/O2/N2體積比為1∶1∶1∶7,35 ml/min),并將原料氣及產物通入氣相色譜中進行在線分析。

圖1 催化劑考評裝置Fig.1 Schematic diagram of the experimental apparatus for propylene epoxidation
使用GC 2060氣相色譜對產物進行定性和定量分析。使用FID 檢測器和RESTEK 毛細柱(0.35 mm×30 m)分析丙烯、乙醛、PO、丙烯醛、丙醛和丙酮,采用TCD 檢測器和TDX-1 填充柱(3 mm×1 m)分析氫氣、氧氣和二氧化碳。
通過歸一化法計算丙烯轉化率、產物生成速率和選擇性,具體計算公式如下所示。
丙烯轉化率:
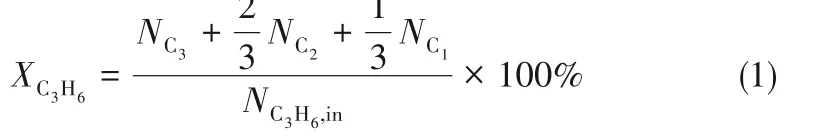
PO生成速率:

PO選擇性:
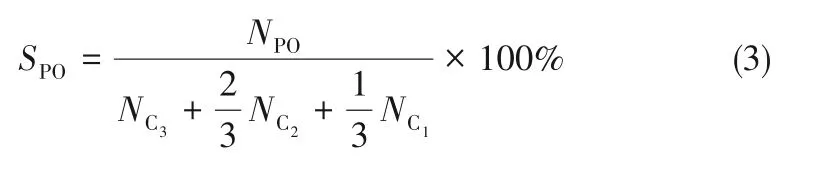
氫效:

2 實驗結果與討論
2.1 TS-1-B-TPAOH與TS-1-B表征
2.1.1 TS-1-B-TPAOH 與TS-1-B 分 子 篩 的 物 相 分析 采用XRD 對TS-1-B 及TS-1-B-TPAOH 分子篩的物相進行了分析,其結果如圖2 所示。從圖中可 知,TS-1-B 及TS-1-B-TPAOH 分 子 篩 在2θ=7.8°,8.8°,23°,23.9°,24.5°處均出現MFI 結構特征峰[18,31],這說明采用TPAOH 溶液對TS-1-B進行二次晶化改性并未破壞TS-1-B 的拓撲結構[26-27]。此外,與TS-1-B 相 比,TS-1-B-TPAOH 分 子 篩 的MFI 結構特征峰強度明顯增強,這說明二次晶化改性提高了TS-1-B的結晶度。這可能是由于TPAOH具有較強的分子篩結構導向能力,在二次晶化處理過程中硅物種發生了溶解-再結晶過程,使得鈦硅分子篩結構得到重整、優化,從而提高了TS-1-B 的結晶度[26]。
2.1.2 TS-1-B-TPAOH 與TS-1-B 分 子 篩 中 鈦 物 種分析 采用UV-Vis 對TS-1-B 及TS-1-B-TPAOH分子篩中鈦物種進行了分析,其結果如圖3 所示。從圖中可以看出,TS-1-B 及TS-1-B-TPAOH 分子篩均在208 nm 左右出現強烈的電子躍遷信號峰,并且這兩個樣品在330 nm 處沒有觀察到明顯的銳鈦礦型二氧化鈦物種吸收峰,這表明所合成的TS-1-B及改性處理后的TS-1-B-TPAOH 分子篩中大部分Ti 以孤立四配位鈦物種的形式存在。可見,采用一定濃度的TPAOH溶液對TS-1-B進行二次晶化處理后鈦物種的組成并未發生明顯變化[18,26,31]。
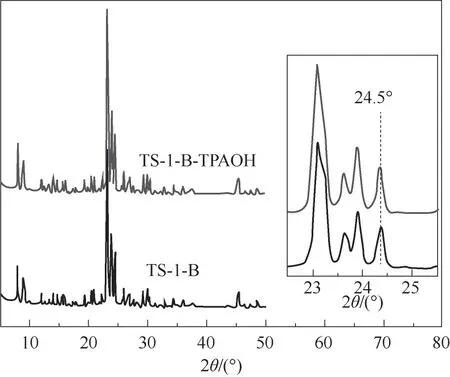
圖2 TS-1-B-TPAOH 與TS-1-B的XRD譜圖Fig.2 XRD patterns of TS-1-B-TPAOH and TS-1-B

圖3 TS-1-B-TPAOH 與TS-1-B的UV-Vis圖Fig.3 UV-Vis spectra of TS-1-B-TPAOH and TS-1-B
2.1.3 TS-1-B-TPAOH 與TS-1-B 分 子篩中硅物種分析 TS-1-B 及TS-1-B-TPAOH 分子篩的FT-IR譜圖如圖4 所示。從圖中可以明顯看出,TS-1-B 及TS-1-B-TPAOH 分子篩均在960 cm-1處出現Si—O—Ti 的不對稱伸縮振動特征峰[18,32],該結果進一步證明鈦原子進入了分子篩骨架,并且TS-1-B 及二次晶化后的TS-1-B-TPAOH 分子篩在960 cm-1處的特征吸收峰強度并沒有明顯的差異,說明二次晶化改性并沒有改變TS-1-B 分子篩表面孤立四配位鈦的含量。此外,TS-1-B 及TS-1-B-TPAOH 分子篩均在550 cm-1處出現MFI 型分子篩五元環的特征峰[18,33],該結果進一步表明二次晶化改性處理后制備的TS-1-B-TPAOH 分子篩仍具有典型的MFI 結構[26-28],這與XRD 結果相一致。另外,TS-1-B 在約1630 cm-1和3450 cm-1處出現兩個特征吸收峰,其中前者歸屬為氫鍵硅羥基特征峰,后者歸屬為氫鍵硅羥基特征峰及吸附于硅羥基上水分子的特征峰[34-36]。然而,二次晶化改性處理后制備的TS-1-B-TPAOH 分子篩在1630 cm-1和3450 cm-1處幾乎觀察不到明顯的特征峰存在,這可能是由于二次晶化改性處理后TS-1-B 的結構得到了重整、優化,從而降低了缺陷位硅羥基的數量[26-28]。

圖4 TS-1-B-TPAOH 與TS-1-B的FT-IR譜圖Fig.4 FT-IR spectra of TS-1-B-TPAOH and TS-1-B
2.1.4 TS-1-B-TPAOH與TS-1-B分子篩的形貌
TS-1-B 與TS-1-B-TPAOH 分子篩的SEM 圖如圖5所示。從圖中可以看出,改性處理前后鈦硅分子篩顆粒大小并未發生明顯的變化,且兩個樣品顆粒比較均勻并且均呈類球形,并不存在無定形物質。此外,從HR-TEM 圖(圖6)可以看出,相比于未改性的TS-1-B 樣品[圖6(a)],改性處理后TS-1-B-TPAOH分子篩[圖6(b)]部分顆粒出現了一些不規則的縫隙。這些縫隙可能是二次晶化過程中部分由納米顆粒堆積形成的TS-1-B進行重新組裝后形成的。
2.1.5 TS-1-B-TPAOH 與TS-1-B 分 子 篩 的 織 構 性質 采用N2物理吸附對TS-1-B 與TS-1-B-TPAOH分子篩的織構性質進行了研究。根據脫附分支數據計算的TS-1-B 與TS-1-B-TPAOH 分子篩BJH 孔徑分布如圖7 所示。從圖中可以看出,相比于未改性TS-1-B 分子篩,采用TPAOH 改性的TS-1-BTPAOH 分子篩在3~20 nm 之間顯示出較寬的介孔分布,這說明采用TPAOH 二次晶化改性TS-1-B 分子篩后產生了一些介孔。

圖5 TS-1-B與TS-1-B-TPAOH 的SEM圖Fig.5 SEM images of TS-1-B and TS-1-B-TPAOH

圖6 TS-1-B與TS-1-B-TPAOH 的HR-TEM圖Fig.6 HR-TEM images of TS-1-B and TS-1-B-TPAOH

圖7 TS-1-B與TS-1-B-TPAOH 的孔徑分布Fig.7 Pore size distributions of TS-1-B and TS-1-B-TPAOH

表1 TS-1-B與TS-1-B-TPAOH分子篩的織構性質Table 1 The structural properties of TS-1-B and TS-1-B-TPAOH
TS-1-B 與TS-1-B-TPAOH 分子篩的比表面積、微孔孔容及總孔容如表1所示。從表中可知,二次晶化改性處理后制備的TS-1-B-TPAOH 分子篩與未改性處理的TS-1-B 分子篩的微孔孔容相差不大,且遠小于傳統焙燒后的鈦硅分子篩TS-1[18,37],說明二次晶化改性處理后的TS-1-B-TPAOH 分子篩微孔孔道仍然被模板劑TPA+所堵塞。這可能是由于填充于未焙燒TS-1 微孔孔道中的TPA+分子良好的穩定性對TS-1-B 分子篩的微孔孔道具有保護作用,從而避免了二次晶化改性處理過程中強堿性TPAOH 溶液對TS-1-B 微孔孔道的腐蝕。此外,從表中還可以看出,二次晶化改性處理后的TS-1-BTPAOH 分子篩的總孔容要高于TS-1-B 分子篩,這也與TS-1-B-TPAOH 分子篩存在一些介孔這一結果相一致。
2.2 TS-1-B-TPAOH 與TS-1-B 分子篩固載金催化劑性能考評
2.2.1 不同載體固載金催化劑上金負載量分析
表2 為TS-1-B-TPAOH 及TS-1-B 固載金催化劑上金負載量的測定結果。從表2 可知,以TS-1-BTPAOH 及TS-1-B 為載體、采用尿素為沉淀劑通過DPU 法制備的金催化劑上金的負載效率均在90%以上,這與文獻報道結果[25]相同。

表2 Au/TS-1-B與Au/TS-1-B-TPAOH催化劑上金負載量Table 2 Au loadings of Au/TS-1-B and Au/TS-1-B-TPAOH catalysts
2.2.2 不同載體固載金催化劑上金顆粒粒徑分析
圖8 為不同金負載量的Au/TS-1-B-TPAOH 催化劑及0.09% Au/TS-1-B 催化劑的HAADF-STEM 圖。從圖中可以看出,0.09%Au/TS-1-B-TPAOH 催化劑與0.09%Au/TS-1-B 催化劑上納米金顆粒的粒徑分布相似且平均粒徑均為2.6 nm 左右。此外,固載量較低的0.08% Au/TS-1-B-TPAOH 催化劑上納米金顆粒的平均粒徑(約2.5 nm)也與0.09% Au/TS-1-B催化劑的相近,這說明在金負載量相差不大時固載于TS-1-B-TPAOH 與TS-1-B 分子篩上的納米金顆粒粒徑沒有明顯差異。
2.2.3 催化劑性能考評 不同金負載量Au/TS-1-B-TPAOH 催化劑與0.09% Au/TS-1-B 催化劑在丙烯氫氧環氧化反應中的丙烯轉化率和PO 生成速率隨時間的變化關系如圖9 所示。從圖中可知,0.09%Au/TS-1-B 催化劑在誘導期之后的丙烯轉化率和PO生成速率隨著反應的進行逐漸降低,約30 h后其丙烯轉化率和PO 生成速率隨著反應的進行不再出現明顯的降低,表明該催化劑在30 h 后達到穩態。0.09% Au/TS-1-B-TPAOH 催化劑在誘導期之后的丙烯轉化率與PO 生成速率隨反應時間變化的曲線可分為兩個階段,其中,該催化劑的丙烯轉化率與PO 生成速率在6~20 h 內基本保持不變,之后該催化劑上丙烯轉化率與PO 生成速率略有降低但很快就達到穩態,并且該催化劑在穩態時的活性明顯高于0.09%Au/TS-1-B 催化劑。此外,0.08%TS-1-B-TPAOH 催化劑在誘導期結束后的丙烯轉化率和PO 生成速率隨著反應的進行也沒有出現明顯的降低現象,并且達到穩態時的丙烯轉化率和PO 生成速率雖低于同樣處于穩態時的0.09% Au/TS-1-B-TPAOH 催化劑,但其活性高于穩態時的0.09%Au/TS-1-B 催化劑。上述催化劑考評結果顯示,相比于0.09% Au/TS-1-B 催化劑,0.09% Au/TS-1-BTPAOH 與0.08%Au/TS-1-B-TPAOH 催化劑顯示出明顯提高的穩定性及穩定的丙烯轉化率和PO 生成速率。
先前研究發現,在Ti—OH 活性中心上生成的PO 在相鄰Ti—OH 或相鄰Si—OH 的作用下能夠發生開環反應形成難以脫附的雙配位丙氧基物種,該物種可進一步轉化為強吸附的羧酸鹽(如甲酸鹽)和碳酸鹽等碳沉積物[20,38-39],這是導致Au-Ti 雙功能催化劑失活的主要原因[20,33-39]。研究還發現,Au-Ti雙功能催化劑表面形成的雙配位丙氧基物種及強吸附的羧酸鹽(如甲酸鹽)和碳酸鹽物種通常在2800~3100 cm-1的紅外波長范圍內出現C—H 鍵的伸縮特征峰[20,38-39]。因此,為比較未改性TS-1-B 與二次晶化改性TS-1-B-TPAOH 固載金催化劑上碳沉積物含量及PO 脫附行為的差異,分析了反應后0.09% Au/TS-1-B 與0.08% Au/TS-1-B-TPAOH 催化劑的FT-IR 譜圖,其結果如圖10 所示。從圖中可以看出,反應后0.08% Au/TS-1-B-TPAOH 催化劑與0.09% Au/TS-1-B 催化劑均在2800~3100 cm-1范圍內出現了三個歸屬為雙配位丙氧基物種與強吸附羧酸鹽(如甲酸鹽)和碳酸鹽物種C—H 鍵的伸縮振動特征峰,并且反應后0.08%Au/TS-1-B-TPAOH催化劑上雙配位丙氧基物種與強吸附羧酸鹽(如甲酸鹽)和碳酸鹽物種C—H 鍵的伸縮振動特征峰強度明顯低于反應后0.09% Au/TS-1-B 催化劑,這預示著前者在反應過程中表面生成的雙配位丙氧基物種與強吸附羧酸鹽(如甲酸鹽)和碳酸鹽物種較少。此外,反應后0.08% Au/TS-1-B-TPAOH 催化劑在1630 cm-1及3450 cm-1處歸屬于氫鍵硅羥基及吸附在分子篩硅羥基上水分子的特征峰強度要低于0.09% Au/TS-1-B 催化劑,這也表明二次晶化改性TS-1-B-TPAOH 固載金催化劑的表面疏水性要高于未改性TS-1-B 固載金催化劑。因此,結合雙配位丙氧基物種與強吸附羧酸鹽(如甲酸鹽)和碳酸鹽物種的形成機理及二次晶化改性TS-1-BTPAOH 固載金催化劑與未改性TS-1-B 固載金催化劑上C—H 鍵伸縮振動特征峰和硅羥基特征峰強度的差異可以推測,相比于未改性TS-1-B 固載的金催化劑,二次晶化改性TS-1-B-TPAOH 固載的金催化劑表面硅羥基含量較低,這有利于促進PO 的脫附,導致催化劑表面生成的雙配位丙氧基物種及碳沉積物較少,這可能是0.08% Au/TS-1-B-TPAOH催化劑相比于0.09%Au/TS-1-B 催化劑表現出較高的穩定性及穩定的丙烯轉化率和PO 生成速率的原因之一。此外,考慮到TS-1-B-TPAOH 的比表面積(24 m2/g)要高于TS-1-B(15 m2/g)。因此,0.08%Au/TS-1-B-TPAOH 催化劑上單位面積的碳沉積物含量也要低于0.09% Au/TS-1-B 催化劑,這可能是0.08%Au/TS-1-B-TPAOH 催化劑相比于0.09%Au/TS-1-B催化劑顯示出較高穩定性的另一個原因。

圖8 0.09%Au/TS-1-B、0.09%Au/TS-1-B-TPAOH 與0.08%Au/TS-1-B-TPAOH 催化劑HAADF-STEM 圖與相應的粒徑分布Fig.8 HAADF-STEM images and Au particle size distribution of 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-B-TPAOH catalysts

圖9 不同載體固載金催化劑上丙烯轉化率與PO生成速率隨時間的變化關系(其中,0.09%Au/TS-1-B催化劑的PO生成速率數據來自本課題組先前報道工作[30])Fig.9 Propylene conversion and PO formation rate over 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-BTPAOH catalysts as a function of time-on-stream(the PO formation rate of 0.09%Au/TS-1-B was taken from previous work[30])

圖10 反應后0.09%Au/TS-1-B與0.08%Au/TS-1-BTPAOH催化劑的FT-IR譜圖Fig.10 FT-IR spetra of spent 0.09%Au/TS-1-B and 0.08%Au/TS-1-B-TPAOH catalysts
催化劑考評結果顯示(圖9),0.08% Au/TS-1-B-TPAOH 催化劑在誘導期之后的丙烯轉化率及PO生成速率低于0.09% Au/TS-1-B-TPAOH 催化劑,但其穩定性要優于金固載量較高的0.09% Au/TS-1-B-TPAOH 催化劑。這可能是由于該催化劑上PO生成速率低于0.09% Au/TS-1-B-TPAOH 催化劑,此時0.08% Au/TS-1-B-TPAOH 催化劑上PO 生成與脫附的良好匹配使得該催化劑相比于0.09% Au/TS-1-B-TPAOH 催化劑顯示出進一步提高的穩定性。因此,根據上述分析也可推測,協調Au-Ti雙功能催化劑上PO 的生成與脫附是提高Au-Ti 雙功能催化劑穩定性的關鍵所在。
除穩定性外,PO 選擇性也是評價Au-Ti 雙功能催化劑性能高低的重要指標之一。先前研究發現Ti活性位點上生成的PO能夠吸附于相鄰Si—OH上并后續發生開環及異構等反應生成副產物[21,33-36],由此可見,促進Ti 活性位點上PO 的脫附有助于提高PO 選擇性。Au/TS-1-B-TPAOH 催化劑與Au/TS-1-B 催化劑在穩態時PO 選擇性如圖11 所示。從圖中可知,0.09%Au/TS-1-B-TPAOH 催化劑在穩態時的PO 選擇性略微高于0.09%Au/TS-1-B 催化劑,這可能是由于0.09% Au/TS-1-B-TPAOH 催化劑表面硅羥基數量較少,這有利于促進PO 在催化劑表面的脫附,因此該催化劑在穩態時丙烯轉化率明顯高于穩態時0.09%Au/TS-1-B 催化劑的情況下,其PO選擇性仍略高于后者。此外,0.08% Au/TS-1-BTPAOH 催化劑在穩態時的PO 選擇性明顯高于穩態時的0.09% Au/TS-1-B-TPAOH 催化劑。考慮到0.08%Au/TS-1-B-TPAOH 催化劑上PO 的生成速率低于0.09%Au/TS-1-B-TPAOH 催化劑,此時0.08%Au/TS-1-B-TPAOH 催化劑上PO 的生成與脫附可能實現了較好的匹配,這可能是該催化劑相比于0.09%Au/TS-1-B-TPAOH 催化劑具有較高PO 選擇性的原因之一。此外,0.08%Au/TS-1-B-TPAOH 催化劑在穩態時的PO 選擇性要明顯高于穩態時的0.09%Au/TS-1-B 催化劑。考慮到該0.08%Au/TS-1-B-TPAOH 催化劑的表面疏水性要高于0.09%Au/TS-1-B 催化劑,這有助于促進PO 在催化劑表面的脫附,這可能是該催化劑在具有較高PO 生成速率與丙烯轉化率情況下還顯示出較高PO 選擇性的主要原因。

圖11 不同載體固載金催化劑在穩態時的PO選擇性與氫效(0.09%Au/TS-1-B催化劑的PO選擇性和氫效數據來自本課題組先前報道工作[30])Fig.11 PO selectivity and hydrogen efficiency over 0.09%Au/TS-1-B,0.09%Au/TS-1-B-TPAOH and 0.08%Au/TS-1-BTPAOH catalysts at steady state(the PO selectivity and hydrogen efficiency of 0.09%Au/TS-1-B was taken from previous work[30])
氫效也是評價Au-Ti雙功能催化劑性能的重要指標之一。Au/TS-1-B-TPAOH 催化劑與Au/TS-1-B 催化劑在穩態時的氫效如圖11 所示。從圖中可知,0.09% Au/TS-1-B-TPAOH 與0.08% Au/TS-1-B-TPAOH 催化劑在穩態時的氫效均明顯高于0.09% Au/TS-1-B 催化劑,這可能是由于以TS-1-B-TPAOH 分子篩為載體固載的金催化劑表面疏水性較高,這有助于促進PO 在催化劑表面的脫附,從而促進了納米金顆粒上氫氣與氧氣反應生成的HOOH 物種與Ti 活性位點反應生成更多的Ti—OOH活性中間體,進而提高了氫氣利用效率[21,33-36]。
2.2.4 反應溫度影響 反應溫度也對Au-Ti雙功能催化劑催化丙烯氫氧環氧化反應具有重要影響。0.08% Au/TS-1-B-TPAOH 催化劑上反應溫度對丙烯氫氧環氧化反應性能的影響如圖12 所示。從圖中可以看出,丙烯的轉化率和PO 生成速率隨著反應溫度的升高而提高,但是PO 選擇性和氫效卻隨著反應溫度的升高而降低。產生這種現象的主要原因可能是隨著反應溫度的升高,氫氣和氧氣在納米金顆粒上生成HOOH 物種的速率和丙烯環氧化的速率均增加,因此使得丙烯轉化率和PO 生成速率也隨反應溫度的升高而提高。然而HOOH 物種的穩定性不高,較高的反應溫度會提高該物種的分解速率,導致大量HOOH 物種在高溫下發生無效分解,并且HOOH 物種分解量的增加也提高了非選擇性氧化反應生成副產物的速率,從而導致PO 選擇性和氫效隨反應溫度的升高而降低[32,40-41]。
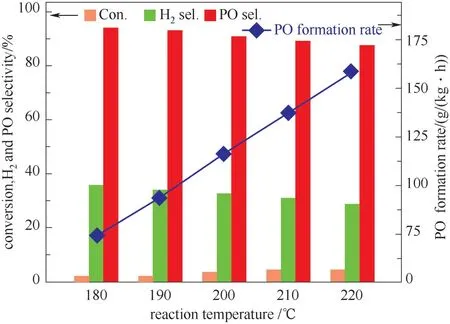
圖12 反應溫度對0.08%Au/TS-1-B-TPAOH 催化劑性能的影響Fig.12 Catalytic performance of 0.08%Au/TS-1-B-TPAOH catalyst as a function of reaction temperature
0.08% Au/TS-1-B-TPAOH 催化劑上副產物選擇性與反應溫度的關系如圖13 所示。從圖中可以看出,該催化劑上生成的副產物有乙醛、丙烯醛、丙醛、丙酮和二氧化碳,其中丙醛是主要副產物。此外,從圖中可以看出,隨著反應溫度的升高,各個副產物的選擇性急劇增加,這預示著升高反應溫度更有利于副產物的生成[32,40,42]。因此,對于丙烯氫氧環氧化反應而言,提高反應溫度反而不利于獲得較高的PO選擇性。

圖13 0.08%Au/TS-1-B-TPAOH 催化劑上反應溫度對副產物選擇性的影響Fig.13 By-products selectivity of 0.08%Au/TS-1-B-TPAOH catalyst with different reaction temperature
主/副產物生成速率與反應溫度的關系如圖14所示,通過Arrhenius 方程擬合得到的主/副產物表觀活化能如表3 所示。從表中可以看出,生成各副產物的活化能均高于生成主產物PO 的活化能,這表明副產物的生成對反應溫度更加敏感[32,40]。此外,0.08% Au/TS-1-B-TPAOH 催化劑上主/副產物的生成活化能位于文獻報道的Au-Ti 催化劑上生成主/副產物活化能的范圍內,說明本文制備的催化劑上丙烯氫氧環氧化反應與文獻報道的催化劑遵從相同的反應機理[32,40,43]。

圖14 0.08%Au/TS-1-B-TPAOH 催化劑上主/副產物生成速率與溫度關系Fig.14 The relationship between the formation rate of PO/byproducts and temperature over 0.08%Au/TS-1-B-TPAOH catalyst
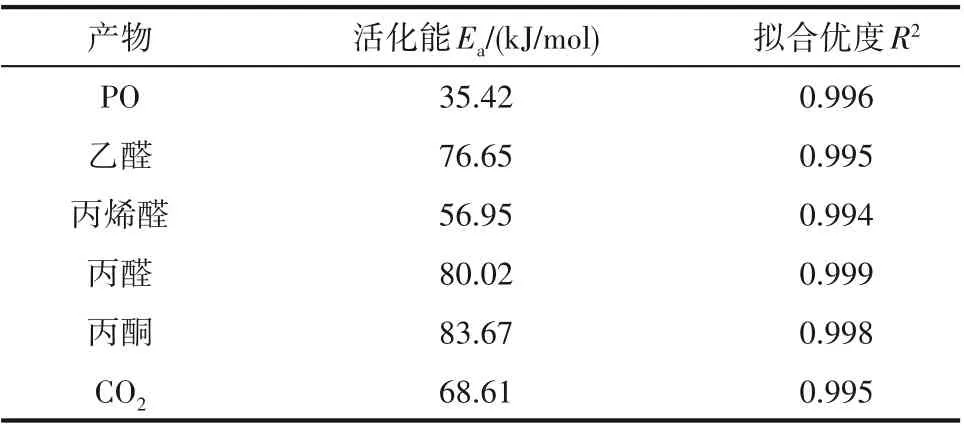
表3 主/副產物表觀活化能Table 3 Apparent activation energy of each by-product by linear regression
3 結 論
本文采用TPAOH對TS-1-B進行二次晶化處理制備了TS-1-B-TPAOH 分子篩,對比研究了DPU法制備的Au/TS-1-B催化劑與Au/TS-1-B參比催化劑在丙烯氫氧環氧化反應中催化行為的差異,并結合FT-IR、HAADF-STEM 等表征手段闡釋了Au/TS-1-B-TPAOH 與Au/TS-1-B 催化劑結構與性能之間的關系。在此基礎上進行了Au/TS-1-B-TPAOH 催化劑的動力學特性分析。結論如下。
(1)TS-1-B 和TS-1-B-TPAOH 分子篩表征結果表明二次晶化改性提高了母體TS-1-B 分子篩的結晶度,減少了分子篩表面缺陷位硅羥基的數量,從而顯著提高了TS-1-B分子篩的表面疏水性。
(2)Au/TS-1-B-TPAOH 催化劑相比于Au/TS-1-B催化劑表現出顯著提高的穩定性及穩定的轉化率、PO 生成速率、PO 選擇性和氫效,并且反應后Au/TS-1-B-TPAOH 催化劑上生成的雙配位丙氧基物種與甲酸鹽等碳沉積物較少,這可歸因于二次晶化改性TS-1-B 分子篩顯著降低了表面硅羥基數量,這有利于抑制PO 吸附于硅羥基上并發生開環、異構等反應生成雙配位丙氧基物種與碳沉積物,從而提高了Au/TS-1-B-TPAOH 催化劑的穩定性與活性。
(3)動力學分析結果顯示,提高反應溫度雖有利于提高丙烯轉化率和PO 生成速率,但會顯著降低PO選擇性和氫效。生成PO的活化能明顯低于生成各副產物的活化能,這表明副反應對反應溫度更加敏感。
符 號 說 明
M——摩爾質量,g/mol
m——催化劑質量,g
N——摩爾流量,mol/h
r——生成速率,g/(kg·h)
S——選擇性,%
X——轉化率,%
下角標
in——進口
out——出口
猜你喜歡
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
汽車觀察(2018年10期)2018-11-06 07:05:26
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
