豇豆中噁唑酰草胺及其代謝物殘留量的超高效液相色譜-串聯質譜法測定
2021-08-06 21:57:49邵輝李娜李輝劉磊林宏芳楊麗靜閆琳程禹劉春艷張玉婷郭永澤
天津農業科學 2021年5期
關鍵詞:測定
邵輝 李娜 李輝 劉磊 林宏芳 楊麗靜 閆琳 程禹 劉春艷 張玉婷 郭永澤

摘? ? 要:本研究建立了豇豆中噁唑酰草胺及其代謝物HPFMPA、HFMPA、6-CBO殘留量的超高效液相色譜-串聯質譜(UPLC-MS/MS)測定方法。采用乙腈提取,PSA凈化,UPLC-MS/MS在多反應離子監測模式(MRM)下測定,外標-標準曲線法定量方法。結果表明,豇豆中添加噁唑酰草胺、HPFMPA、HFMPA、6-CBO濃度水平為0.005 ~1.000 mg·kg-1,平均回收率在75%~97%,相對標準偏差在5%~13%,定量限在0.005~0.010 mg·kg-1。方法可同時測定噁唑酰草胺及其代謝物,準確度高,靈敏度好,特異性強,前處理操作簡單,適用于豇豆中噁唑酰草胺及其代謝物殘留量測定。
關鍵詞:豇豆;噁唑酰草胺;代謝物;超高效液相色譜-串聯質譜法;測定
中圖分類號:S643.4; O657.63? ? ? ? ? ? ? ? ?文獻標識碼:A? ? ? ? ? ? ? ? DOI 編碼:10.3969/j.issn.1006-6500.2021.05.016
Abstract: An accurate UPLC-MS/MS method for the determination of metamifop and its metabolites HPFMPA,HFMPA,6-CBO in cowpea was developed. Cowpea samples were extracted with acetonitrile, cleaned up by PSA ,and finally determinated by UPLC-MS/MS under multiple reaction monitoring (MRM) mode. The results showed that the concentration levels of metamifop, HPFMPA, HFMPA and 6-CBO added to cowpea were 0.005-1.000 mg·kg-1, the average recoveries of metamifop and its metabolites were between 75% and 97% with the RSDs between 5% and 13%, the limits of quantitation were between 0.005 mg·kg-1 and 0.010 mg·kg-1. The method is qualitative, quantitative, sensitive, specific, simple, suitable for determination of metamifop and its metabolites in cowpea.
Key words: cowpea; metamifop; metabolites; UPLC-MS/MS; determination
噁唑酰草胺,英文名稱為metamifop,化學名稱為(R)-2-{ (4-氯-1,3-苯并噁唑-2-基氧)苯氧基}-2′-氟-N-甲基丙酰替苯胺,是由韓國化工技術研究院開發的芳氧苯氧丙酸酯類除草劑,屬ACC酶抑制劑,能抑制植物脂肪酸的合成,主要用于移栽和直播稻田防除一年生禾本科雜草,有廣泛的可混性,并有望用于其他作物,是一個很有發展前景的除草劑[1-3]。噁唑酰草胺在田間施用以后,會降解為有毒理學意義的代謝物N-(2-氟苯基)-2-(4-羥基苯氧基)-N-甲基丙酰胺(HPFMPA)、N-(2-氟苯基)-2-羥基-N-甲基丙酰胺(HFMPA)以及6-氯-2-苯噁唑啉酮(6-CBO)。目前國內外對于噁唑酰草胺及其代謝物殘留量的測定方法研究已有報道,如大米中噁唑酰草胺及其代謝物殘留量的高效液相色譜測定法(HPLC)[4-6],水和糧谷中噁唑酰草胺殘留量的液相色譜-串聯質譜測定法(LC/MS/MS)[7-9],大米和水中噁唑酰草胺殘留量的液相色譜測定法[10-14],如水稻、水和土中噁唑酰草胺及其代謝物殘留量的液相色譜-串聯質譜法[15-16]等。但是,尚未有豇豆中噁唑酰草胺及其代謝物殘留量的超高效液相色譜-串聯質譜(UPLC-MS/MS)測定法報道。
基于此,本研究建立了這一方法,為測定豇豆中噁唑酰草胺及其代謝物殘留量和噁唑酰草胺在豇豆上登記使用提供技術參考。
1 材料和方法
1.1 試驗材料
1.1.1 儀器設備 Acquity UPLC/Xevo TQS micro超高效液相色譜-串聯質譜儀(美國Waters公司)、T50勻漿機(德國IKA公司)、0.01 g電子天平(天津天馬衡基儀器有限公司)、CK2000垂直振蕩器(托摩根生物科技有限公司)、3-18KS高速離心機(德國Sigma公司)、QL-901渦旋振蕩器(江蘇海門其林貝爾儀器制造有限公司)、Milli-Q純水儀(美國Millipore公司)、100 μL移液槍(德國eppendorf公司)。
1.1.2 試劑? 乙腈(分析純,天津市康科德科技有限公司)、鹽酸(分析純,天津市風船化學試劑科技有限公司)、氯化鈉(分析純,天津市風船化學試劑科技有限公司)、乙腈(色譜純,德國默克公司)、甲酸(色譜純,德國默克公司)、超純水(Milli-Q純水儀制備)、PSA(顆粒直徑為60 μm,上海安譜實驗科技股份有限公司)。
噁唑酰草胺(100 mg·L-1,天津阿爾塔科技有限公司)、噁唑酰草胺代謝物HPFMPA(99.1%,杭州宇昊化工科技有限公司)、噁唑酰草胺代謝物HFMPA(98.9%,國家農藥質量監督檢驗測試中心(沈陽))、噁唑酰草胺代謝物6-CBO(99.5%,美國Dr.Ehrenstorfer GmbH公司)。
1.2 測定方法
1.2.1 樣品前處理 稱取5.00 g樣品(精確至0.01 g)于50 mL離心管中,加入5 mL 1 mol·L-1鹽酸,搖勻,再加25 mL乙腈、3.00 g氯化鈉,垂直振蕩5 min,4 000 r·min-1離心5 min。取上清液1.5 mL,加入25 mg PSA,渦旋振蕩1 min,4 000 r·min-1離心5 min,取上清液,過0.22 μm濾膜于進樣小瓶中,待測。
1.2.2 儀器條件 色譜柱:Acquity UPLC BEH C18柱 (50×2.1 mm,1.8 μm);流動相:A為乙腈,B為0.1%甲酸水溶液,體積比:A/B=85/15,等度洗脫;流速:0.3 mL·min-1;洗脫時間:3 min;柱溫:33 ℃;進樣量:5 μL。
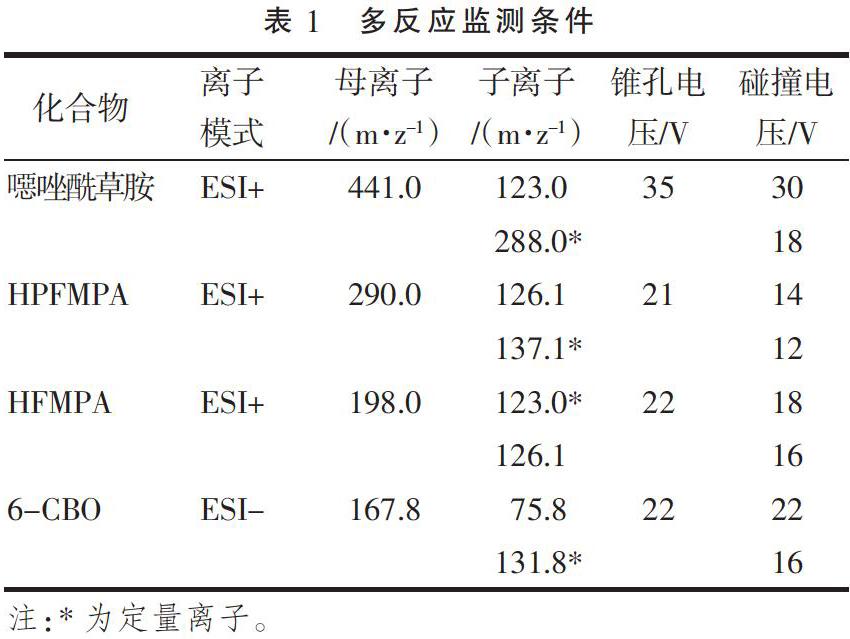
離子源:電子噴霧離子源;毛細管電壓:3.0 KV;脫溶劑溫度:450 ℃;離子源溫度:150 ℃;錐孔氣流速:900 L·h-1;脫溶劑氣流速:50 L·h-1。掃描方式:多反應監測(MRM),條件見表1。
1.2.3 定性 在相同儀器條件下進行樣品測定,如果檢出的色譜峰的保留時間與標準樣品相同,并且在扣除背景后的樣品質譜圖中,目標化合物選擇的離子均出現,而且同一檢測批次,對同一化合物,樣品中目標化合物的離子豐度比與質量濃度相當的基質標準溶液相比一致,則可判斷樣品中存在目標農藥。
1.2.4 定量 外標-基質匹配標準曲線法定量殘留量計算公式如下:
式中,R為樣品中農藥殘留量(mg·kg-1);C為根據標準曲線計算的檢測樣品中的農藥殘留濃度(mg·L-1);V為定容體積(mL);W為代表檢測樣品的樣品質量(g)。
殘留量之和計算公式如下:
R噁唑酰草胺殘留量之和=R噁唑酰草胺 + RHPFMPA ×440.9/289.3+ RHFMPA ×440.9/197.2+ R6-CBO×440.9/169.6(2)
式中,R噁唑酰草胺殘留量之和為噁唑酰草胺殘留量之和(mg·kg-1);R噁唑酰草胺為噁唑酰草胺殘留量(mg·kg-1);RHPFMPA為HPFMPA殘留量(mg·kg-1);RHFMPA-HFMPA殘留量(mg·kg-1);R6-CBO-6-CBO殘留量(mg·kg-1)。
2 結果與分析
2.1 質譜條件的確定
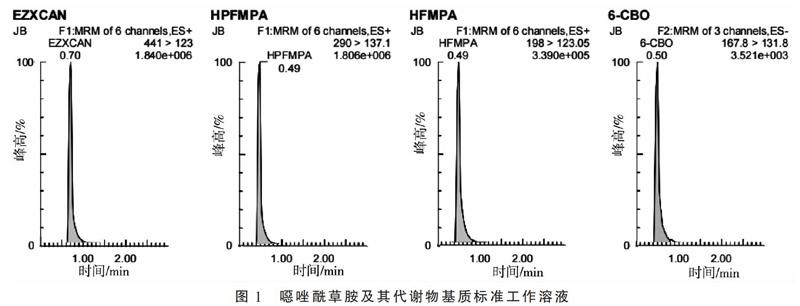
配制噁唑酰草胺及其代謝物溶劑標準工作溶液,根據其理化性質,在合適的離子模式(正離子模式或負離子模式)下對進行母離子掃描,結合其分子量,確定母離子大小,然后對錐孔電壓進行優化,確定母離子響應最大時的錐孔電壓。接下來對母離子施加碰撞電壓進行子離子掃描,找到響應較大且穩定的2個作為定性子離子,再分別對兩個子離子進行碰撞電壓的優化,找到子離子響應最大時的碰撞電壓,最后選擇響應較高的子離子作為定量子離子,使儀器靈敏度達到最佳。同樣的方法,得到噁唑酰草胺、HPFMPA、HFMPA、6-CBO的母離子、錐孔電壓、子離子、碰撞電壓,見表1,基質標準工作溶液譜圖(圖1)。
2.2 前處理方法的確定
本研究采用乙腈提取,乙腈可有效提取大多數樣品中大多數農藥及其代謝物殘留,且提取出來的影響定性定量的脂類物質較少,更為重要的是,通過加入氯化鈉,乙腈相可與水相分層,達到去除水溶性雜質的目的。再加入PSA作為凈化吸附劑,可有效去除樣品中影響農殘測定的色素、酸類、糖類等物質,降低了基質對信號響應的抑制,凈化效果良好,提高了方法靈敏度,保護液相色譜-串聯質譜儀免受污染和堵塞,可延長儀器壽命并提高穩定性,而且噁唑酰草胺及其代謝物的回收率均可以達到要求。
2.3 標準曲線線性關系分析
用豇豆基質空白溶液稀釋,配制噁唑酰草胺/HPFMPA/HFMPA/6-CBO混合標準工作溶液系列濃度0.001/0.0005/0.001/0.001 mg·L-1,0.002/0.001/0.002/0.002 mg·L-1,0.01/0.01/0.01/0.01 mg·L-1,0.05/0.05/0.05/0.05 mg·L-1,0.2/0.2/0.2/0.2 mg·L-1。在上述液相色譜-串聯質譜條件下進樣,以化合物濃度為橫坐標,以對應的峰面積為縱坐標作標準曲線,得噁唑酰草胺線性方程為y=1 431 219.2x-181.5,r = 0.999 7;HPFMPA線性方程為y=1 329 521.9x+ 306.1,r=0.999 9;HFMPA線性方程為y=344 752.0x -167.7,r=0.999 7;6-CBO線性方程為y=103 942.3x-107.0,r=0.999 8。線性關系均良好,標準曲線見圖2。
2.4 方法的特異性
本方法采用多反應監測(MRM)掃描模式,先對母離子進行掃描,再對母離子產生的子離子進行掃描,而且選擇兩對母離子-子離子反應掃描來定性定量,保證了基本只對目標化合物有響應,對雜質有良好的區分能力,可有效地排除假陽性結果。
2.5 方法的靈敏度
方法的靈敏度以定量限(LOQ)表示。根據實測,本方法噁唑酰草胺、HPFMPA、HFMPA、6-CBO的定量限分別為0.01,0.005,0.01,0.01 mg·kg-1,國內外均未制定噁唑酰草胺在豇豆中的最大殘留限量(MRL)值,定量限足以達到農藥殘留量測定的要求。
2.6 方法的準確度和精密度
添加噁唑酰草胺0.01,0.05,1.00 mg·kg-1,平均回收率分別為75%,84%,77%,相對標準偏差分別為6%,8%,5%。添加HPFMPA 0.005,0.050,1.000 mg·kg-1,平均回收率分別為90%,83%,91%,相對標準偏差分別為12%,12%,10%。添加HFMPA 0.01,
0.05,1.00 mg·kg-1,平均回收率分別為90%,90%,82%,相對標準偏差分別為13%,7%,12%。添加6-CBO 0.01,0.05,1.00 mg·kg-1,平均回收率分別為84%,97%,85%,相對標準偏差分別為7%,13%,8%。平均回收率和相對標準偏差均滿足農藥殘留量測定的要求,具體見表2。
3 結 論
不同類型的樣品所含成分不同,樣品中成分可影響待測物的提取效率和儀器響應,在一類樣品上建立了檢測方法,在其他類樣品上不一定適用,因此還需要驗證、改進才能使用。已有文獻報道的液相色譜-串聯質譜儀測定噁唑酰草胺及其代謝物殘留量的方法,涉及到的樣品有水、土壤、糙米、稻殼、秸稈。豇豆含水量高、葉綠素含量高、極性雜質較多,和前人研究過的樣品大有不同,本研究采用乙腈提取、PSA凈化、超高效液相色譜-串聯質譜儀測定、外標法定量,建立了豇豆中噁唑酰草胺及其代謝物殘留量的超高效液相色譜-串聯質譜儀測定方法,拓寬了樣品檢測類型范圍。方法可同時測定噁唑酰草胺及其代謝物,準確度高,靈敏度好,特異性強,在實際樣品測定中準確高效,可為豇豆中噁唑酰草胺及其代謝物殘留量測定提供技術支撐。
參考文獻:
[1] 馬國蘭, 劉都才, 劉雪源, 等. 五氟磺草胺等6種除草劑對直播稻田高齡稗草的生物活性及田間控制效果[J]. 植物保護, 2014, 40(3): 204-208, 214.
[2] 黃誼. 噁唑酰草胺10%EC防除直播稻田禾本科雜草研究[J]. 農業災害研究, 2015, 5(1): 11-13, 41.
[3] LEICHTER C A, THOMPSON N, JOHNSON B R, et al. The high potency of ME-5343 to aphids is due to a unique mechanism of action[J]. Pesticide Biochemistry and Physiology, 2013, 107(2): 169-176.
[4] 羅婧, 施海燕, 彭文濤, 等. 噁唑酰草胺及其代謝物的殘留分析方法[J]. 江蘇農業學報, 2010, 26(1): 187-191.
[5] 孔德洋, 石利利, 單正軍, 等. 惡唑酰草胺及其代謝物殘留的加速溶劑萃取-凝膠滲透色譜凈化-液相色譜測定[J]. 環境化學, 2010, 29(4): 734-738.
[6] 王點點, 陳源, 宋寧慧, 等. 10%噁唑酰草胺可濕性粉劑在稻田環境中的殘留動態[J]. 農藥, 2012, 51(11): 818-821.
[7] 劉雁雨, 魏京華, 張燕, 等. 超高效液相色譜—串聯質譜法同時檢測水中五氟磺草胺和噁唑酰草胺[J]. 農藥科學與管理, 2018, 39(5): 51-55.
[8] 唐俗, 袁定帥, 孫林, 等. 液相色譜-串聯質譜法測定糧谷中的噁唑酰草胺[J]. 食品工業, 2019, 40(3): 319-321.
[9] 張月. 超高效液相色譜-串聯質譜法測定水稻中噁唑酰草胺殘留量[J]. 中國農學通報, 2020, 36(9): 127-131.
[10] 孔德洋, 石利利, 單正軍, 等. 噁唑酰草胺在稻田中的殘留及消解動態[J]. 生態與農村環境學報, 2011, 27(5): 104-107.
[11] 羅婧, 彭文濤, 金雅慧, 等. 噁唑酰草胺的高效液相色譜分析[J]. 農藥, 2009, 48(3): 191-192.
[12] 王雪, 侯志廣, 郭剛, 等. 噁唑酰草胺的光解動力學研究[J]. 中國農學通報, 2014, 30(1): 312-315.
[13] 楊麗莉, 王雪, 崔強, 等. 300W高壓汞燈下惡唑酰草胺在不同表面活性劑水溶液中的光解動態[J]. 吉林農業, 2015(23): 66-66.
[14] 王雪, 楊麗莉, 崔強, 等. 不同表面活性劑溶液下惡唑酰草胺的水解動態[J]. 吉林農業, 2015(23): 69.
[15] 陳國峰, 尤紅梅, 滕瑤, 等. UPLC-MS/MS法測定水稻中噁唑酰草胺和氰氟草酯及其代謝物的殘留及膳食風險評估[J]. 中國稻米, 2021, 27(2): 57-62.
[16] 豆葉枝, 李菊穎, 何健, 等. 分散固相萃取-液相色譜質譜法測定水稻和多環境介質中的噁唑酰草胺及其代謝產物[J]. 環境化學, 2020, 39(10): 2693-2701.
猜你喜歡
農機使用與維修(2016年12期)2017-01-17 17:30:33
安徽農學通報(2016年24期)2017-01-12 02:10:04
科學與財富(2016年29期)2016-12-27 16:45:09
科學與財富(2016年29期)2016-12-27 16:43:09
科學與財富(2016年29期)2016-12-27 16:42:10
綠色科技(2016年20期)2016-12-27 16:04:51
東方教育(2016年3期)2016-12-14 20:35:31
中國科技博覽(2016年13期)2016-07-13 01:51:35
中國科技博覽(2016年11期)2016-05-06 04:30:49
中國科技博覽(2016年10期)2016-04-29 03:36:52
